BioBB DNA Command Line Help
Generic usage:
biobb_command [-h] --config CONFIG --input_file(s) <input_file(s)> --output_file <output_file>
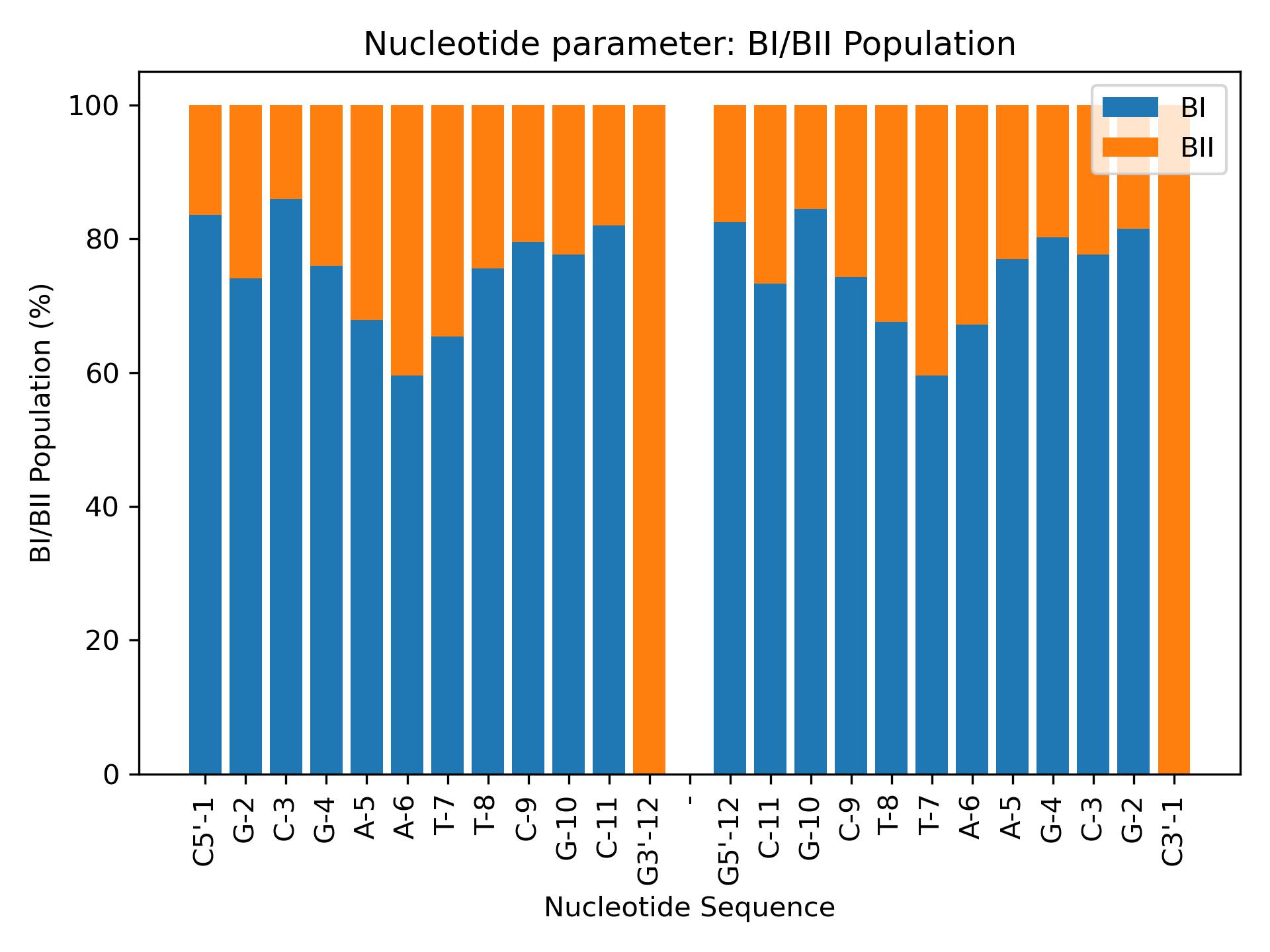
Bipopulations
Calculate BI/BII populations from epsilon and zeta parameters.
Get help
Command:
bipopulations -h
usage: bipopulations [-h] [--config CONFIG] --input_epsilC_path INPUT_EPSILC_PATH --input_epsilW_path INPUT_EPSILW_PATH --input_zetaC_path INPUT_ZETAC_PATH --input_zetaW_path INPUT_ZETAW_PATH --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Calculate BI/BII populations.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_epsilC_path INPUT_EPSILC_PATH
Helical parameter <epsilC> input ser file path. Accepted formats: ser.
--input_epsilW_path INPUT_EPSILW_PATH
Helical parameter <epsilW> input ser file path. Accepted formats: ser.
--input_zetaC_path INPUT_ZETAC_PATH
Helical parameter <zetaC> input ser file path. Accepted formats: ser.
--input_zetaW_path INPUT_ZETAW_PATH
Helical parameter <zetaW> input ser file path. Accepted formats: ser.
--output_csv_path OUTPUT_CSV_PATH
Path to output csv file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output jpg file. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_epsilC_path (string): Path to .ser file for helical parameter ‘epsilC’. File type: input. Sample file. Accepted formats: SER
input_epsilW_path (string): Path to .ser file for helical parameter ‘epsilW’. File type: input. Sample file. Accepted formats: SER
input_zetaC_path (string): Path to .ser file for helical parameter ‘zetaC’. File type: input. Sample file. Accepted formats: SER
input_zetaW_path (string): Path to .ser file for helical parameter ‘zetaW’. File type: input. Sample file. Accepted formats: SER
output_csv_path (string): Path to .csv file where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
sequence (string): (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option)..
seqpos (array): (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
sequence: CGCGAATTCGCG
Command line
bipopulations --config config_bipopulations.yml --input_epsilC_path canal_output_epsilC.ser --input_epsilW_path canal_output_epsilW.ser --input_zetaC_path canal_output_zetaC.ser --input_zetaW_path canal_output_zetaW.ser --output_csv_path bipop_ref.csv --output_jpg_path bipop_ref.jpg
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG"
}
}
Command line
bipopulations --config config_bipopulations.json --input_epsilC_path canal_output_epsilC.ser --input_epsilW_path canal_output_epsilW.ser --input_zetaC_path canal_output_zetaC.ser --input_zetaW_path canal_output_zetaW.ser --output_csv_path bipop_ref.csv --output_jpg_path bipop_ref.jpg
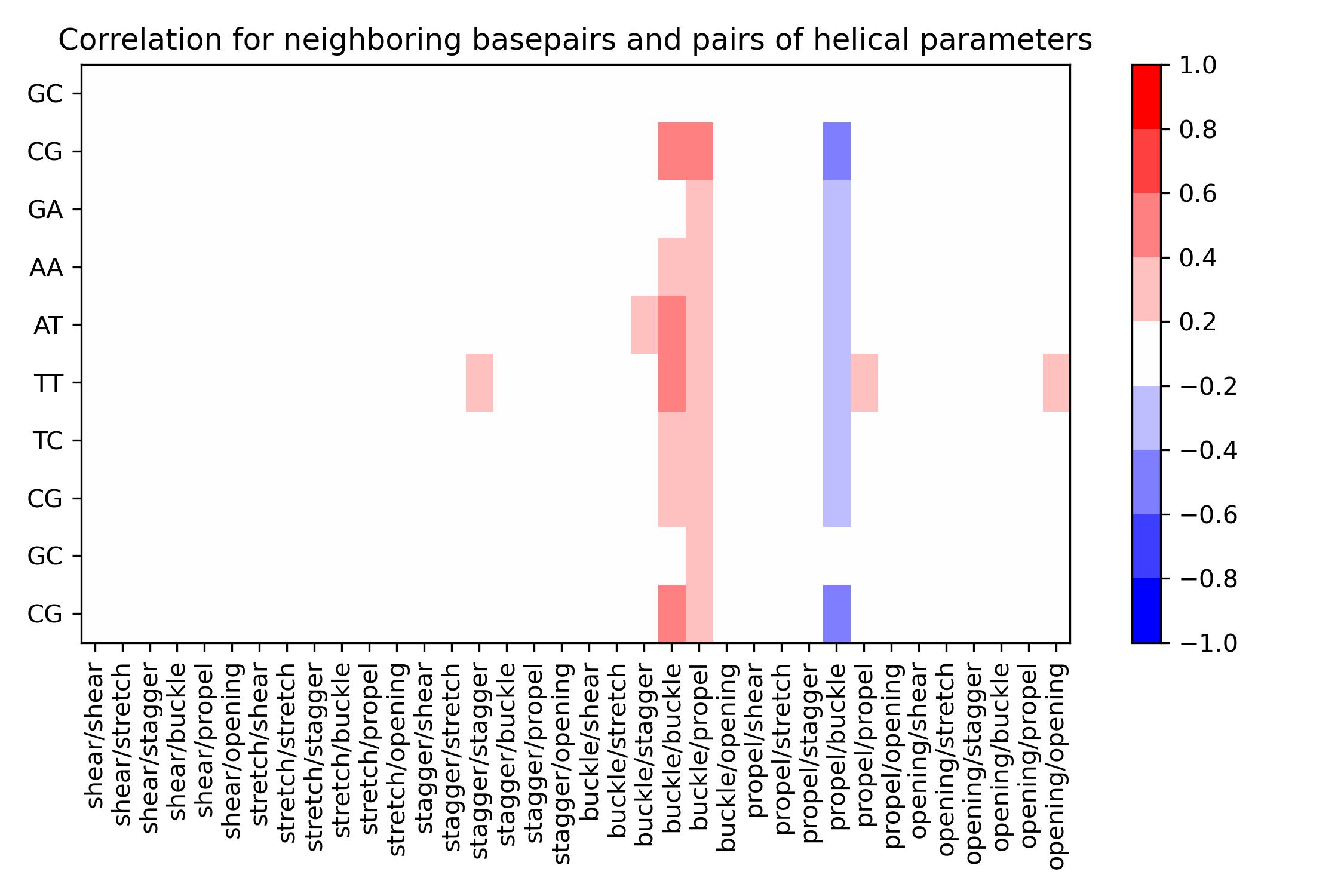
Intrabpcorr
Calculate correlation between all intra-base pairs of a single sequence and for a single helical parameter.
Get help
Command:
intrabpcorr -h
usage: intrabpcorr [-h] [--config CONFIG] --input_filename_shear INPUT_FILENAME_SHEAR --input_filename_stretch INPUT_FILENAME_STRETCH --input_filename_stagger INPUT_FILENAME_STAGGER --input_filename_buckle INPUT_FILENAME_BUCKLE --input_filename_propel INPUT_FILENAME_PROPEL --input_filename_opening INPUT_FILENAME_OPENING --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Load .ser file from Canal output and calculate correlation between base pairs of the corresponding sequence.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_filename_shear INPUT_FILENAME_SHEAR
Path to ser file for helical parameter shear. Accepted formats: ser.
--input_filename_stretch INPUT_FILENAME_STRETCH
Path to ser file for helical parameter stretch. Accepted formats: ser.
--input_filename_stagger INPUT_FILENAME_STAGGER
Path to ser file for helical parameter stagger. Accepted formats: ser.
--input_filename_buckle INPUT_FILENAME_BUCKLE
Path to ser file for helical parameter buckle. Accepted formats: ser.
--input_filename_propel INPUT_FILENAME_PROPEL
Path to ser file for helical parameter propel. Accepted formats: ser.
--input_filename_opening INPUT_FILENAME_OPENING
Path to ser file for helical parameter opening. Accepted formats: ser.
--output_csv_path OUTPUT_CSV_PATH
Path to output file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output plot. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_filename_shear (string): Path to .ser file with data for helical parameter ‘shear’. File type: input. Sample file. Accepted formats: SER
input_filename_stretch (string): Path to .ser file with data for helical parameter ‘stretch’. File type: input. Sample file. Accepted formats: SER
input_filename_stagger (string): Path to .ser file with data for helical parameter ‘stagger’. File type: input. Sample file. Accepted formats: SER
input_filename_buckle (string): Path to .ser file with data for helical parameter ‘buckle’. File type: input. Sample file. Accepted formats: SER
input_filename_propel (string): Path to .ser file with data for helical parameter ‘propel’. File type: input. Sample file. Accepted formats: SER
input_filename_opening (string): Path to .ser file with data for helical parameter ‘opening’. File type: input. Sample file. Accepted formats: SER
output_csv_path (string): Path to directory where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
sequence (string): (None) Nucleic acid sequence for the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option)..
seqpos (array): (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
sequence: CGCGAATTCGCG
Command line
intrabpcorr --config config_intrabpcorr.yml --input_filename_shear canal_output_shear.ser --input_filename_stretch canal_output_stretch.ser --input_filename_stagger canal_output_stagger.ser --input_filename_buckle canal_output_buckle.ser --input_filename_propel canal_output_propel.ser --input_filename_opening canal_output_opening.ser --output_csv_path intra_bpcorr_ref.csv --output_jpg_path intra_bpcorr_ref.jpg
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG"
}
}
Command line
intrabpcorr --config config_intrabpcorr.json --input_filename_shear canal_output_shear.ser --input_filename_stretch canal_output_stretch.ser --input_filename_stagger canal_output_stagger.ser --input_filename_buckle canal_output_buckle.ser --input_filename_propel canal_output_propel.ser --input_filename_opening canal_output_opening.ser --output_csv_path intra_bpcorr_ref.csv --output_jpg_path intra_bpcorr_ref.jpg
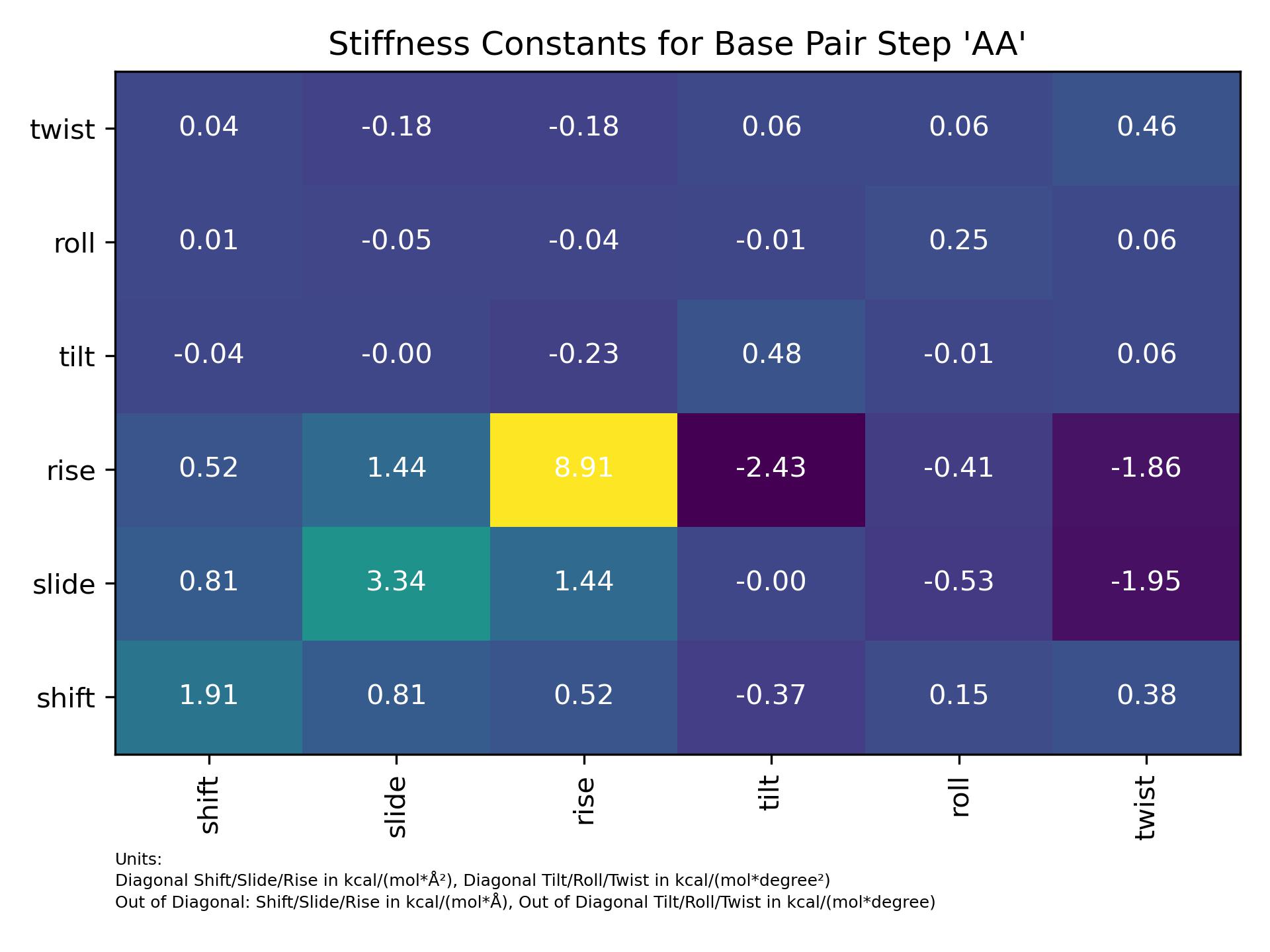
Basepair_stiffness
Calculate stiffness constants matrix between all six helical parameters for a single base pair step.
Get help
Command:
basepair_stiffness -h
usage: basepair_stiffness [-h] [--config CONFIG] --input_filename_shift INPUT_FILENAME_SHIFT --input_filename_slide INPUT_FILENAME_SLIDE --input_filename_rise INPUT_FILENAME_RISE --input_filename_tilt INPUT_FILENAME_TILT --input_filename_roll INPUT_FILENAME_ROLL --input_filename_twist INPUT_FILENAME_TWIST --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Calculate stiffness constants matrix between all six helical parameters for a single base pair step.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_filename_shift INPUT_FILENAME_SHIFT
Path to csv file with shift inputs. Accepted formats: csv.
--input_filename_slide INPUT_FILENAME_SLIDE
Path to csv file with slide inputs. Accepted formats: csv.
--input_filename_rise INPUT_FILENAME_RISE
Path to csv file with rise inputs. Accepted formats: csv.
--input_filename_tilt INPUT_FILENAME_TILT
Path to csv file with tilt inputs. Accepted formats: csv.
--input_filename_roll INPUT_FILENAME_ROLL
Path to csv file with roll inputs. Accepted formats: csv.
--input_filename_twist INPUT_FILENAME_TWIST
Path to csv file with twist inputs. Accepted formats: csv.
--output_csv_path OUTPUT_CSV_PATH
Path to output covariance data file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output covariance data file. Accepted formats: csv.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_filename_shift (string): Path to csv file with data for helical parameter ‘shift’. File type: input. Sample file. Accepted formats: CSV
input_filename_slide (string): Path to csv file with data for helical parameter ‘slide’. File type: input. Sample file. Accepted formats: CSV
input_filename_rise (string): Path to csv file with data for helical parameter ‘rise’. File type: input. Sample file. Accepted formats: CSV
input_filename_tilt (string): Path to csv file with data for helical parameter ‘tilt’. File type: input. Sample file. Accepted formats: CSV
input_filename_roll (string): Path to csv file with data for helical parameter ‘roll’. File type: input. Sample file. Accepted formats: CSV
input_filename_twist (string): Path to csv file with data for helical parameter ‘twist’. File type: input. Sample file. Accepted formats: CSV
output_csv_path (string): Path to directory where stiffness matrix file is saved as a csv file. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to directory where stiffness heatmap image is saved as a jpg file. File type: output. Sample file. Accepted formats: JPG
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
KT (number): (0.592186827) Value of Boltzmann temperature factor..
scaling (array): ([1, 1, 1, 10.6, 10.6, 10.6]) Values by which to scale stiffness. Positions correspond to helical parameters in the order: shift, slide, rise, tilt, roll, twist..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
remove_tmp: false
Command line
basepair_stiffness --config config_basepair_stiffness.yml --input_filename_shift series_shift_AA.csv --input_filename_slide series_slide_AA.csv --input_filename_rise series_rise_AA.csv --input_filename_tilt series_tilt_AA.csv --input_filename_roll series_roll_AA.csv --input_filename_twist series_twist_AA.csv --output_csv_path stiffbp_ref.csv --output_jpg_path stiffbp_ref.jpg
JSON
Common config file
{
"properties": {
"remove_tmp": false
}
}
Command line
basepair_stiffness --config config_basepair_stiffness.json --input_filename_shift series_shift_AA.csv --input_filename_slide series_slide_AA.csv --input_filename_rise series_rise_AA.csv --input_filename_tilt series_tilt_AA.csv --input_filename_roll series_roll_AA.csv --input_filename_twist series_twist_AA.csv --output_csv_path stiffbp_ref.csv --output_jpg_path stiffbp_ref.jpg
Dna_averages
Load .ser file for a given helical parameter and read each column corresponding to a base calculating average over each one.
Get help
Command:
dna_averages -h
usage: dna_averages [-h] [--config CONFIG] --input_ser_path INPUT_SER_PATH --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Load helical parameter file and calculate average values for each base pair.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_ser_path INPUT_SER_PATH
Helical parameter input ser file path. Accepted formats: ser.
--output_csv_path OUTPUT_CSV_PATH
Path to output csv file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output jpg file. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_ser_path (string): Path to .ser file for helical parameter. File is expected to be a table, with the first column being an index and the rest the helical parameter values for each base/basepair. File type: input. Sample file. Accepted formats: SER
output_csv_path (string): Path to .csv file where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
sequence (string): (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option)..
helpar_name (string): (Optional) helical parameter name..
stride (integer): (1000) granularity of the number of snapshots for plotting time series..
seqpos (array): (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
seqpos:
- 4
- 5
- 6
sequence: CGCGAATTCGCG
stride: 1
Command line
dna_averages --config config_dna_averages.yml --input_ser_path canal_output_shift.ser --output_csv_path shift_avg.csv --output_jpg_path shift_avg.jpg
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG",
"seqpos": [
4,
5,
6
],
"stride": 1
}
}
Command line
dna_averages --config config_dna_averages.json --input_ser_path canal_output_shift.ser --output_csv_path shift_avg.csv --output_jpg_path shift_avg.jpg
Biobb_canal
Wrapper for the Canal executable that is part of the Curves+ software suite.
Get help
Command:
biobb_canal -h
usage: biobb_canal [-h] [--config CONFIG] --input_cda_file INPUT_CDA_FILE --output_zip_path OUTPUT_ZIP_PATH [--input_lis_file INPUT_LIS_FILE]
Execute Canal from the Curves+ software suite.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
--input_lis_file INPUT_LIS_FILE
lis input file from Curves+ output. Accepted formats: lis.
required arguments:
--input_cda_file INPUT_CDA_FILE
cda input file from Curves+ output. Accepted formats: cda.
--output_zip_path OUTPUT_ZIP_PATH
Filename for .zip file with Canal output. Accepted formats: zip.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_cda_file (string): Input cda file, from Cur+ output. File type: input. Sample file. Accepted formats: CDA
input_lis_file (string): Input lis file, from Cur+ output. File type: input. Sample file. Accepted formats: LIS
output_zip_path (string): zip filename for output files. File type: output. Sample file. Accepted formats: ZIP
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
bases (string): (None) sequence of bases to be searched for in the I/P data (default is blank, meaning no specified sequence)..
itst (integer): (0) Iteration start index..
itnd (integer): (0) Iteration end index..
itdel (integer): (1) Iteration delimiter..
lev1 (integer): (0) Lower base level limit (i.e. base pairs) used for analysis..
lev2 (integer): (0) Upper base level limit used for analysis. If lev1 > 0 and lev2 = 0, lev2 is set to lev1 (i.e. analyze lev1 only). If lev1=lev2=0, lev1 is set to 1 and lev2 is set to the length of the oligmer (i.e. analyze all levels)..
nastr (string): (NA) character string used to indicate missing data in .ser files..
cormin (number): (0.6) minimal absolute value for printing linear correlation coefficients between pairs of analyzed variables..
series (boolean): (False) if True then output spatial or time series data. Only possible for the analysis of single structures or single trajectories..
histo (boolean): (False) if True then output histogram data..
corr (boolean): (False) if True than output linear correlation coefficients between all variables..
sequence (string): (Optional) sequence of the first strand of the corresponding DNA fragment, for each .cda file. If not given it will be parsed from .lis file..
binary_path (string): (Canal) Path to Canal executable, otherwise the program wil look for Canal executable in the binaries folder..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
corr: true
histo: true
sequence: CGCGAATTCGCG
series: true
Command line
biobb_canal --config config_biobb_canal.yml --input_cda_file curves_output.cda --input_lis_file input.lis --output_zip_path canal_output.zip
JSON
Common config file
{
"properties": {
"series": true,
"histo": true,
"corr": true,
"sequence": "CGCGAATTCGCG"
}
}
Command line
biobb_canal --config config_biobb_canal.json --input_cda_file curves_output.cda --input_lis_file input.lis --output_zip_path canal_output.zip
Canal_unzip
Tool for extracting biobb_canal output files.
Get help
Command:
canal_unzip -h
usage: canal_unzip [-h] [--config CONFIG] --input_zip_file INPUT_ZIP_FILE --output_path OUTPUT_PATH [--output_list_path OUTPUT_LIST_PATH]
Tool for extracting biobb_canal output files.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
--output_list_path OUTPUT_LIST_PATH
Text file with a list of all Canal output files contained within input_zip_file. Accepted formats: txt.
required arguments:
--input_zip_file INPUT_ZIP_FILE
Zip file with Canal output files. Accepted formats: zip.
--output_path OUTPUT_PATH
Canal output file contained within input_zip_file. Accepted formats: ser, his, cor.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_zip_file (string): Zip file with Canal output files. File type: input. Sample file. Accepted formats: ZIP
output_path (string): Canal output file contained within input_zip_file. File type: output. Sample file. Accepted formats: SER, HIS, COR
output_list_path (string): Text file with a list of all Canal output files contained within input_zip_file. File type: output. Sample file. Accepted formats: TXT
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
type (string): (None) Type of file. .
helpar_name (string): (None) Helical parameter name, only for ‘series’ and ‘histo’ types. .
correlation (string): (None) Correlation indexes separated by underscore (ie ‘98_165’), only for ‘corr’ type..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
helpar_name: alphaC
type: histo
Command line
canal_unzip --config config_canal_unzip.yml --input_zip_file canal_output.zip --output_path canal_unzip_output.ser --output_list_path canal_unzip_output.txt
JSON
Common config file
{
"properties": {
"type": "histo",
"helpar_name": "alphaC"
}
}
Command line
canal_unzip --config config_canal_unzip.json --input_zip_file canal_output.zip --output_path canal_unzip_output.ser --output_list_path canal_unzip_output.txt
Biobb_canion
Wrapper for the Canion executable that is part of the Curves+ software suite.
Get help
Command:
biobb_canion -h
usage: biobb_canion [-h] [--config CONFIG] --input_cdi_path INPUT_CDI_PATH --input_afr_path INPUT_AFR_PATH --input_avg_struc_path INPUT_AVG_STRUC_PATH [--output_zip_path OUTPUT_ZIP_PATH]
Execute Canion form the Curves+ software suite.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
--output_zip_path OUTPUT_ZIP_PATH
Filename to give to output files. Accepted formats: zip.
required arguments:
--input_cdi_path INPUT_CDI_PATH
Ion position data file. Accepted formats: cdi.
--input_afr_path INPUT_AFR_PATH
Helical axis frames data. Accepted formats: afr.
--input_avg_struc_path INPUT_AVG_STRUC_PATH
Average DNA conformation fike file. Accepted formats: pdb.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_cdi_path (string): Trajectory input file. File type: input. Sample file. Accepted formats: CDI
input_afr_path (string): Helical axis frames corresponding to the input conformation to be analyzed. File type: input. Sample file. Accepted formats: AFR
input_avg_struc_path (string): Average DNA conformation. File type: input. Sample file. Accepted formats: PDB
output_zip_path (string): Filename for .zip files containing Canion output files. File type: output. Sample file. Accepted formats: ZIP
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
bases (string): (None) Sequence of bases to be analyzed (default is blank, meaning no specified sequence)..
type (string): () Ions (or atoms) to be analyzed. Options are ‘Na+’, ‘K’, ‘K+’, ‘Cl’, ‘Cl-’, ‘CL’, ‘P’, ‘C1’, ‘NH1’, ‘NH2’, ‘NZ’, ‘1’ for all cations, ‘-1’ for all anions, ‘0’ for neutral species or ‘*’ for all available data..
dlow (number): (0.0) Select starting segment of the oglimer to analyze. If both dhig and dlow are 0, entire oglimer is analyzed..
dhig (number): (0.0) Select ending segment of the oglimer to analyze, being the maximum value the total number of base pairs in the oligomer. If both dhig and dlow are 0, entire oglimer is analyzed..
rlow (number): (0.0) Minimal distances from the helical axis taken into account in the analysis..
rhig (number): (0.0) Maximal distances from the helical axis taken into account in the analysis..
alow (number): (0.0) Minimal angle range to analyze..
ahig (number): (360.0) Maximal angle range to analyze..
itst (integer): (0) Number of first snapshot to be analyzed..
itnd (integer): (0) Number of last snapshot to be analyzed..
itdel (integer): (1) Spacing between analyzed snapshots..
rmsf (boolean): (False) If set to True uses the combination of the helical ion parameters and an average helical axis to map the ions into Cartesian space and then calculates their average position (pdb output) and their root mean square fluctuation values (rmsf output). A single pass rmsf algorithm to make this calculation possible with a single read of the trajectory file. This option is generally used for solute atoms and not for solvent molecules or ions..
circ (boolean): (False) If set to True, minicircles are analyzed..
binary_path (string): (Canion) Path to Canion executable, otherwise the program wil look for Canion executable in the binaries folder..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
dhig: 7.2
dlow: 5.8
rhig: 18
rlow: 0
type: K+
Command line
biobb_canion --config config_biobb_canion.yml --input_cdi_path THGA_K.cdi --input_afr_path THGA.afr --input_avg_struc_path THGA_avg.pdb --output_zip_path canion_output.zip
JSON
Common config file
{
"properties": {
"dlow": 5.8,
"dhig": 7.2,
"type": "K+",
"rlow": 0,
"rhig": 18
}
}
Command line
biobb_canion --config config_biobb_canion.json --input_cdi_path THGA_K.cdi --input_afr_path THGA.afr --input_avg_struc_path THGA_avg.pdb --output_zip_path canion_output.zip
Intrahpcorr
Calculate correlation between helical parameters for a single intra-base pair.
Get help
Command:
intrahpcorr -h
usage: intrahpcorr [-h] [--config CONFIG] --input_filename_shear INPUT_FILENAME_SHEAR --input_filename_stretch INPUT_FILENAME_STRETCH --input_filename_stagger INPUT_FILENAME_STAGGER --input_filename_buckle INPUT_FILENAME_BUCKLE --input_filename_propel INPUT_FILENAME_PROPEL --input_filename_opening INPUT_FILENAME_OPENING --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Load helical parameter file and save base data individually.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_filename_shear INPUT_FILENAME_SHEAR
Path to csv file with inputs. Accepted formats: csv.
--input_filename_stretch INPUT_FILENAME_STRETCH
Path to csv file with inputs. Accepted formats: csv.
--input_filename_stagger INPUT_FILENAME_STAGGER
Path to csv file with inputs. Accepted formats: csv.
--input_filename_buckle INPUT_FILENAME_BUCKLE
Path to csv file with inputs. Accepted formats: csv.
--input_filename_propel INPUT_FILENAME_PROPEL
Path to csv file with inputs. Accepted formats: csv.
--input_filename_opening INPUT_FILENAME_OPENING
Path to csv file with inputs. Accepted formats: csv.
--output_csv_path OUTPUT_CSV_PATH
Path to output file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output file. Accepted formats: csv.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_filename_shear (string): Path to .csv file with data for helical parameter ‘shear’. File type: input. Sample file. Accepted formats: CSV
input_filename_stretch (string): Path to .csv file with data for helical parameter ‘stretch’. File type: input. Sample file. Accepted formats: CSV
input_filename_stagger (string): Path to .csv file with data for helical parameter ‘stagger’. File type: input. Sample file. Accepted formats: CSV
input_filename_buckle (string): Path to .csv file with data for helical parameter ‘buckle’. File type: input. Sample file. Accepted formats: CSV
input_filename_propel (string): Path to .csv file with data for helical parameter ‘propeller’. File type: input. Sample file. Accepted formats: CSV
input_filename_opening (string): Path to .csv file with data for helical parameter ‘opening’. File type: input. Sample file. Accepted formats: CSV
output_csv_path (string): Path to directory where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
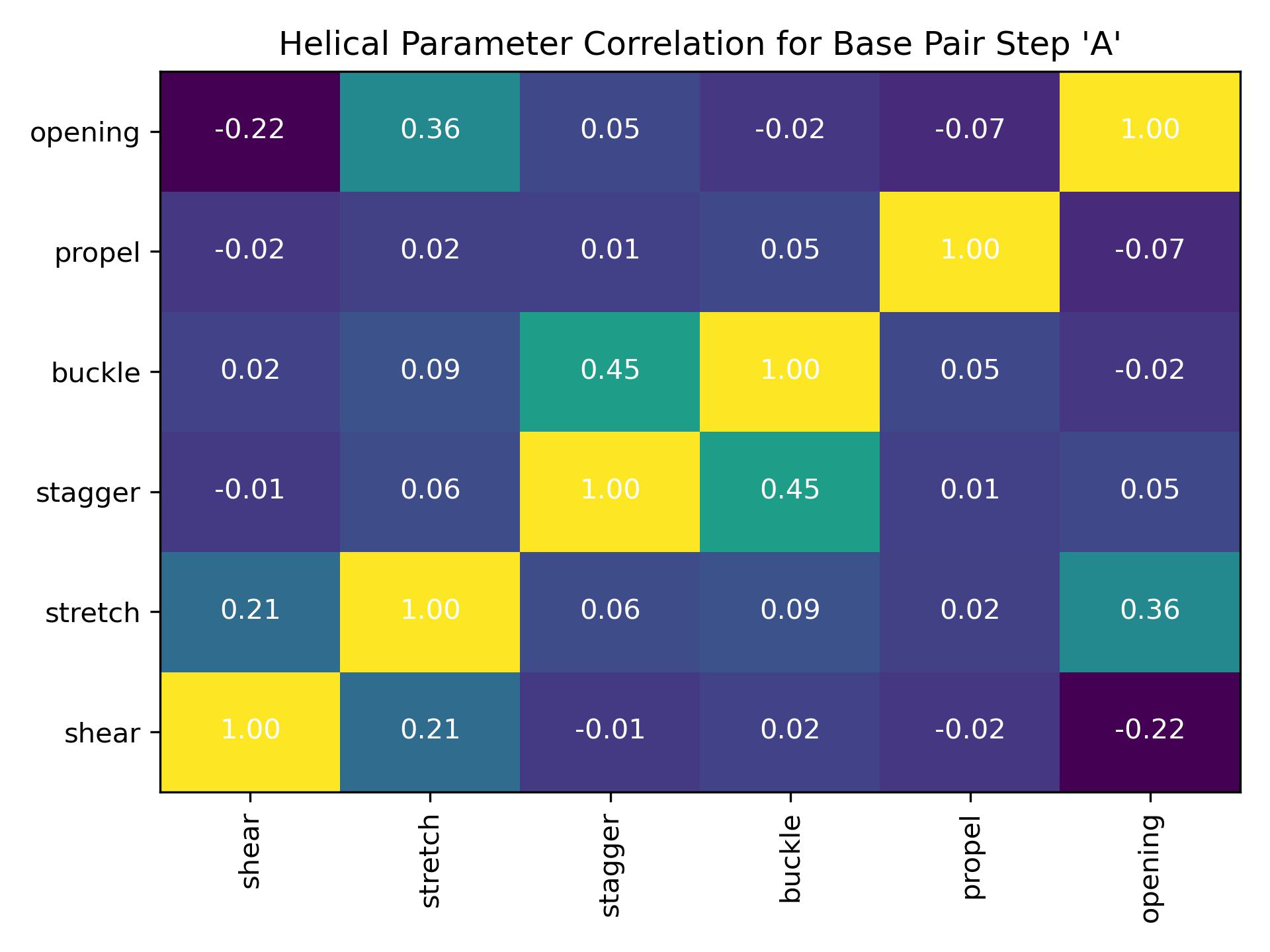{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
base (string): (None) Name of base analyzed..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
remove_tmp: false
Command line
intrahpcorr --config config_intrahpcorr.yml --input_filename_shear series_shear_A.csv --input_filename_stretch series_stretch_A.csv --input_filename_stagger series_stagger_A.csv --input_filename_buckle series_buckle_A.csv --input_filename_propel series_propel_A.csv --input_filename_opening series_opening_A.csv --output_csv_path intra_hpcorr_ref.csv --output_jpg_path intra_hpcorr_ref.jpg
JSON
Common config file
{
"properties": {
"remove_tmp": false
}
}
Command line
intrahpcorr --config config_intrahpcorr.json --input_filename_shear series_shear_A.csv --input_filename_stretch series_stretch_A.csv --input_filename_stagger series_stagger_A.csv --input_filename_buckle series_buckle_A.csv --input_filename_propel series_propel_A.csv --input_filename_opening series_opening_A.csv --output_csv_path intra_hpcorr_ref.csv --output_jpg_path intra_hpcorr_ref.jpg
Intraseqcorr
Calculate correlation between all intra-base pairs of a single sequence and for a single helical parameter.
Get help
Command:
intraseqcorr -h
usage: intraseqcorr [-h] [--config CONFIG] --input_ser_path INPUT_SER_PATH --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Load .ser file from Canal output and calculate correlation between base pairs of the corresponding sequence.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_ser_path INPUT_SER_PATH
Path to ser file with inputs. Accepted formats: ser.
--output_csv_path OUTPUT_CSV_PATH
Path to output file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output plot. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_ser_path (string): Path to .ser file with data for single helical parameter. File type: input. Sample file. Accepted formats: SER
output_csv_path (string): Path to directory where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
sequence (string): (None) Nucleic acid sequence for the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option)..
helpar_name (string): (None) helical parameter name to add to plot title..
seqpos (array): (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
sequence: CGCGAATTCGCG
Command line
intraseqcorr --config config_intraseqcorr.yml --input_ser_path canal_output_buckle.ser --output_csv_path intra_seqcorr_buckle.csv --output_jpg_path intra_seqcorr_buckle.jpg
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG"
}
}
Command line
intraseqcorr --config config_intraseqcorr.json --input_ser_path canal_output_buckle.ser --output_csv_path intra_seqcorr_buckle.csv --output_jpg_path intra_seqcorr_buckle.jpg
Dna_bimodality
Determine binormality/bimodality from a helical parameter series dataset.
Get help
Command:
dna_bimodality -h
usage: dna_bimodality [-h] [--config CONFIG] --input_csv_file INPUT_CSV_FILE [--input_zip_file INPUT_ZIP_FILE] --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Determine binormality/bimodality from a helical parameter dataset.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
--input_zip_file INPUT_ZIP_FILE
Path to zip file containing csv input files. Accepted formats: zip.
required arguments:
--input_csv_file INPUT_CSV_FILE
Path to csv file with data. Accepted formats: csv.
--output_csv_path OUTPUT_CSV_PATH
Filename and/or path of output csv file.
--output_jpg_path OUTPUT_JPG_PATH
Filename and/or path of output jpg file.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_csv_file (string): Path to .csv file with helical parameter series. If
input_zip_fileis passed, this should be just the filename of the .csv file inside .zip. File type: input. Sample file. Accepted formats: CSVinput_zip_file (string): .zip file containing the
input_csv_file.csv file. File type: input. Sample file. Accepted formats: ZIPoutput_csv_path (string): Path to .csv file where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
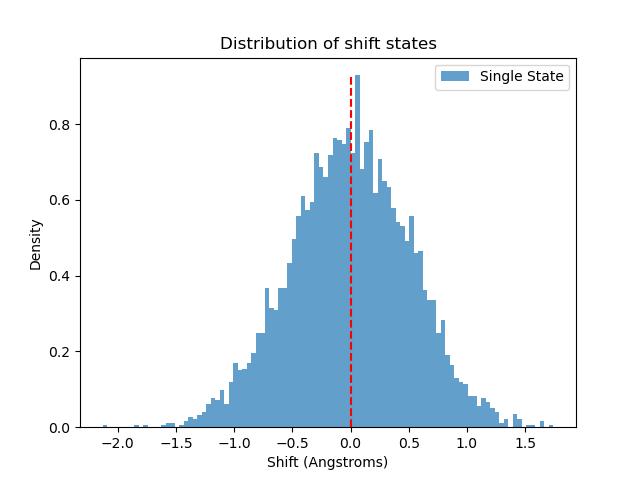{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
helpar_name (string): (Optional) helical parameter name..
confidence_level (number): (5.0) Confidence level for Byes Factor test (in percentage)..
max_iter (integer): (400) Number of maximum iterations for EM algorithm..
tol (number): (1e-05) Tolerance value for EM algorithm..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory.1.
YAML
Common config file
properties:
confidence_level: 5.0
helpar_name: shift
max_iter: 400
tol: 1.0e-05
Command line
dna_bimodality --config config_dna_bimodality.yml --input_csv_file series_shift_AT.csv --input_zip_file input.zip --output_csv_path AT_shift_bimod.csv --output_jpg_path AT_shift_bimod.jpg
JSON
Common config file
{
"properties": {
"helpar_name": "shift",
"confidence_level": 5.0,
"max_iter": 400,
"tol": 1e-05
}
}
Command line
dna_bimodality --config config_dna_bimodality.json --input_csv_file series_shift_AT.csv --input_zip_file input.zip --output_csv_path AT_shift_bimod.csv --output_jpg_path AT_shift_bimod.jpg
Puckering
Calculate Puckering from phase parameters.
Get help
Command:
puckering -h
usage: puckering [-h] [--config CONFIG] --input_phaseC_path INPUT_PHASEC_PATH --input_phaseW_path INPUT_PHASEW_PATH --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Calculate North/East/West/South distribution of sugar puckering backbone torsions.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_phaseC_path INPUT_PHASEC_PATH
Helical parameter <alphaC> input ser file path. Accepted formats: ser.
--input_phaseW_path INPUT_PHASEW_PATH
Helical parameter <alphaW> input ser file path. Accepted formats: ser.
--output_csv_path OUTPUT_CSV_PATH
Path to output csv file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output jpg file. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_phaseC_path (string): Path to .ser file for helical parameter ‘phaseC’. File type: input. Sample file. Accepted formats: SER
input_phaseW_path (string): Path to .ser file for helical parameter ‘phaseW’. File type: input. Sample file. Accepted formats: SER
output_csv_path (string): Path to .csv file where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
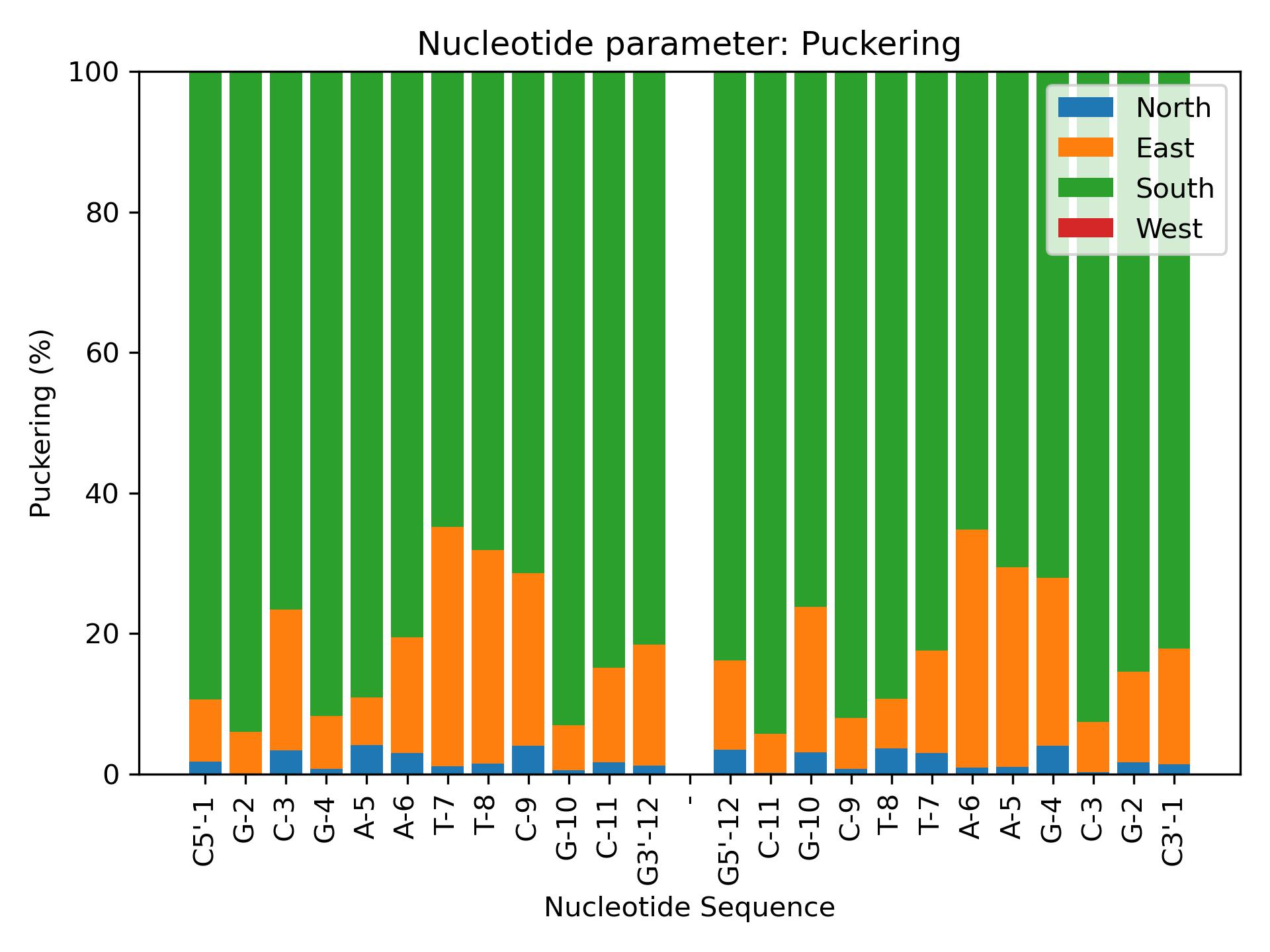{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
sequence (string): (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option)..
stride (integer): (1000) granularity of the number of snapshots for plotting time series..
seqpos (array): (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
sequence: CGCGAATTCGCG
Command line
puckering --config config_puckering.yml --input_phaseC_path canal_output_phaseC.ser --input_phaseW_path canal_output_phaseW.ser --output_csv_path puckering_ref.csv --output_jpg_path puckering_ref.jpg
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG"
}
}
Command line
puckering --config config_puckering.json --input_phaseC_path canal_output_phaseC.ser --input_phaseW_path canal_output_phaseW.ser --output_csv_path puckering_ref.csv --output_jpg_path puckering_ref.jpg
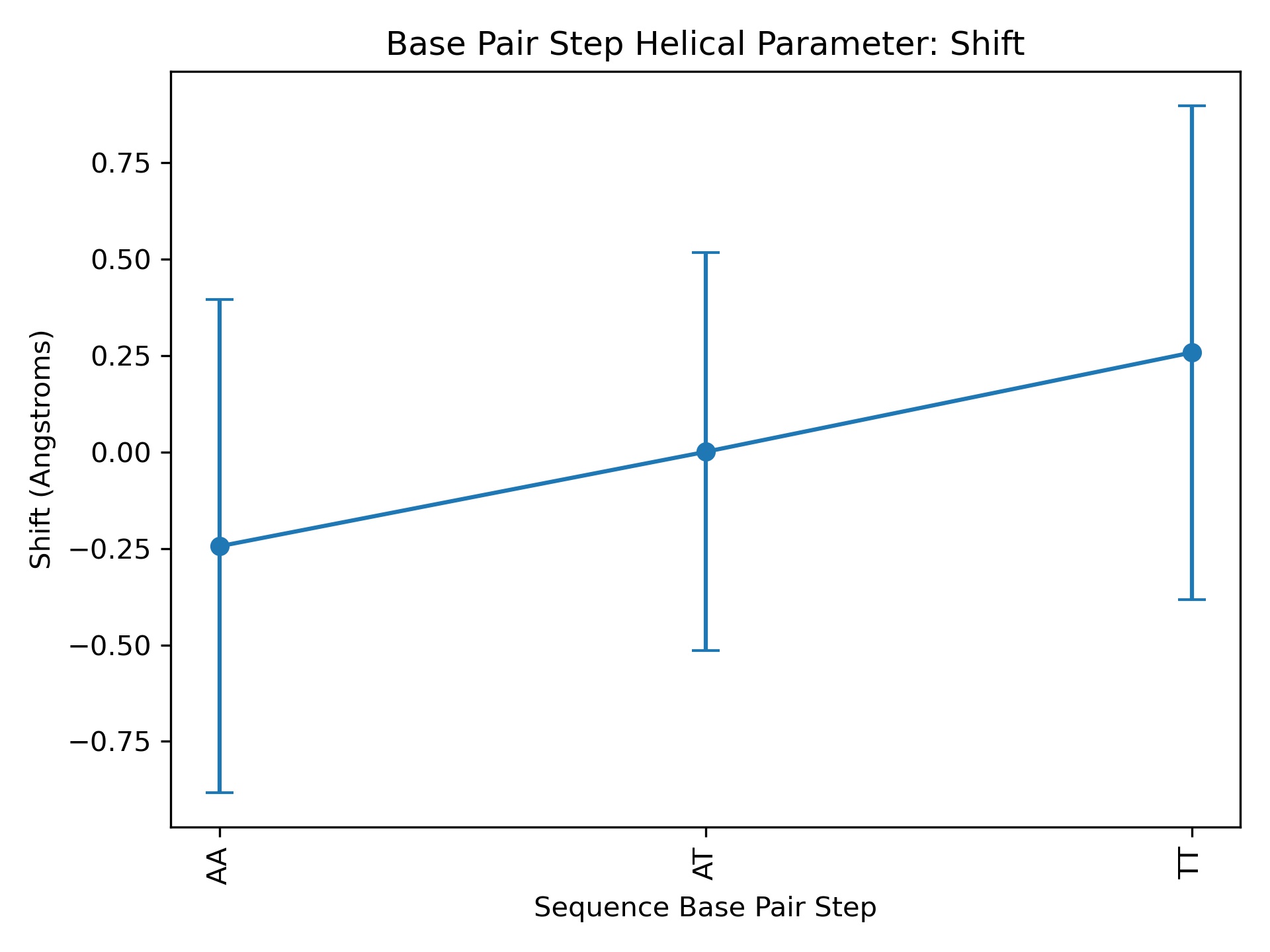
Dna_timeseries
Created time series and histogram plots for each base pair from a helical parameter series file.
Get help
Command:
dna_timeseries -h
usage: dna_timeseries [-h] [--config CONFIG] --input_ser_path INPUT_SER_PATH --output_zip_path OUTPUT_ZIP_PATH
Created time series and histogram plots for each base pair from a helical parameter series file.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_ser_path INPUT_SER_PATH
Helical parameter input ser file path. Accepted formats: ser.
--output_zip_path OUTPUT_ZIP_PATH
Path to output directory.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_ser_path (string): Path to .ser file for helical parameter. File is expected to be a table, with the first column being an index and the rest the helical parameter values for each base/basepair. File type: input. Sample file. Accepted formats: SER
output_zip_path (string): Path to output .zip files where data is saved. File type: output. Sample file. Accepted formats: ZIP
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
sequence (string): (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the usecols option)..
bins (integer): (None) Bins for histogram. Parameter has same options as matplotlib.pyplot.hist..
helpar_name (string): (None) Helical parameter name. It must match the name of the helical parameter in the .ser input file. .
stride (integer): (1000) granularity of the number of snapshots for plotting time series..
seqpos (array): (None) list of sequence positions (columns indices starting by 1) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
seqpos:
- 4
- 5
sequence: CGCGAATTCGCG
Command line
dna_timeseries --config config_dna_timeseries.yml --input_ser_path canal_output_shift.ser --output_zip_path timeseries_output.zip
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG",
"seqpos": [
4,
5
]
}
}
Command line
dna_timeseries --config config_dna_timeseries.json --input_ser_path canal_output_shift.ser --output_zip_path timeseries_output.zip
Interbpcorr
Calculate correlation between all base pairs of a single sequence and for a single helical parameter.
Get help
Command:
interbpcorr -h
usage: interbpcorr [-h] [--config CONFIG] --input_filename_shift INPUT_FILENAME_SHIFT --input_filename_slide INPUT_FILENAME_SLIDE --input_filename_rise INPUT_FILENAME_RISE --input_filename_tilt INPUT_FILENAME_TILT --input_filename_roll INPUT_FILENAME_ROLL --input_filename_twist INPUT_FILENAME_TWIST --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Load .ser file from Canal output and calculate correlation between base pairs of the corresponding sequence.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_filename_shift INPUT_FILENAME_SHIFT
Path to ser file for helical parameter shift. Accepted formats: ser.
--input_filename_slide INPUT_FILENAME_SLIDE
Path to ser file for helical parameter slide. Accepted formats: ser.
--input_filename_rise INPUT_FILENAME_RISE
Path to ser file for helical parameter rise. Accepted formats: ser.
--input_filename_tilt INPUT_FILENAME_TILT
Path to ser file for helical parameter tilt. Accepted formats: ser.
--input_filename_roll INPUT_FILENAME_ROLL
Path to ser file for helical parameter roll. Accepted formats: ser.
--input_filename_twist INPUT_FILENAME_TWIST
Path to ser file for helical parameter twist. Accepted formats: ser.
--output_csv_path OUTPUT_CSV_PATH
Path to output file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output plot. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_filename_shift (string): Path to .ser file with data for helical parameter ‘shift’. File type: input. Sample file. Accepted formats: SER
input_filename_slide (string): Path to .ser file with data for helical parameter ‘slide’. File type: input. Sample file. Accepted formats: SER
input_filename_rise (string): Path to .ser file with data for helical parameter ‘rise’. File type: input. Sample file. Accepted formats: SER
input_filename_tilt (string): Path to .ser file with data for helical parameter ‘tilt’. File type: input. Sample file. Accepted formats: SER
input_filename_roll (string): Path to .ser file with data for helical parameter ‘roll’. File type: input. Sample file. Accepted formats: SER
input_filename_twist (string): Path to .ser file with data for helical parameter ‘twist’. File type: input. Sample file. Accepted formats: SER
output_csv_path (string): Path to directory where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
sequence (string): (None) Nucleic acid sequence for the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option)..
seqpos (array): (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
sequence: CGCGAATTCGCG
Command line
interbpcorr --config config_interbpcorr.yml --input_filename_shift canal_output_shift.ser --input_filename_slide canal_output_slide.ser --input_filename_rise canal_output_rise.ser --input_filename_tilt canal_output_tilt.ser --input_filename_roll canal_output_roll.ser --input_filename_twist canal_output_twist.ser --output_csv_path inter_bpcorr_ref.csv --output_jpg_path inter_bpcorr_ref.jpg
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG"
}
}
Command line
interbpcorr --config config_interbpcorr.json --input_filename_shift canal_output_shift.ser --input_filename_slide canal_output_slide.ser --input_filename_rise canal_output_rise.ser --input_filename_tilt canal_output_tilt.ser --input_filename_roll canal_output_roll.ser --input_filename_twist canal_output_twist.ser --output_csv_path inter_bpcorr_ref.csv --output_jpg_path inter_bpcorr_ref.jpg
Dna_timeseries_unzip
Tool for extracting dna_timeseries output files.
Get help
Command:
dna_timeseries_unzip -h
usage: dna_timeseries_unzip [-h] [--config CONFIG] --input_zip_file INPUT_ZIP_FILE --output_path_csv OUTPUT_PATH_CSV --output_path_jpg OUTPUT_PATH_JPG [--output_list_path OUTPUT_LIST_PATH]
Tool for extracting dna_timeseries output files.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
--output_list_path OUTPUT_LIST_PATH
Text file with a list of all dna_timeseries output files contained within input_zip_file. Accepted formats: txt.
required arguments:
--input_zip_file INPUT_ZIP_FILE
Zip file with dna_timeseries output files. Accepted formats: zip.
--output_path_csv OUTPUT_PATH_CSV
dna_timeseries output csv file contained within input_zip_file. Accepted formats: csv.
--output_path_jpg OUTPUT_PATH_JPG
dna_timeseries output jpg file contained within input_zip_file. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_zip_file (string): Zip file with dna_timeseries output files. File type: input. Sample file. Accepted formats: ZIP
output_path_csv (string): dna_timeseries output csv file contained within input_zip_file. File type: output. Sample file. Accepted formats: CSV
output_path_jpg (string): dna_timeseries output jpg file contained within input_zip_file. File type: output. Sample file. Accepted formats: JPG
output_list_path (string): Text file with a list of all dna_timeseries output files contained within input_zip_file. File type: output. Sample file. Accepted formats: TXT
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
type (string): (None) Type of analysis, series or histogram. .
parameter (string): (None) Type of parameter. .
sequence (string): (None) Nucleic acid sequence used for generating dna_timeseries output file..
index (integer): (1) Base pair index in the parameter ‘sequence’, starting from 1..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
index: 5
parameter: shift
sequence: CGCGAATTCGCG
type: hist
Command line
dna_timeseries_unzip --config config_dna_timeseries_unzip.yml --input_zip_file timeseries_output.zip --output_path_csv dna_timeseries_unzip.csv --output_path_jpg dna_timeseries_unzip.jpg --output_list_path dna_timeseries_unzip.txt
JSON
Common config file
{
"properties": {
"type": "hist",
"parameter": "shift",
"sequence": "CGCGAATTCGCG",
"index": 5
}
}
Command line
dna_timeseries_unzip --config config_dna_timeseries_unzip.json --input_zip_file timeseries_output.zip --output_path_csv dna_timeseries_unzip.csv --output_path_jpg dna_timeseries_unzip.jpg --output_list_path dna_timeseries_unzip.txt
Interseqcorr
Calculate correlation between all base pairs of a single sequence and for a single helical parameter.
Get help
Command:
interseqcorr -h
usage: interseqcorr [-h] [--config CONFIG] --input_ser_path INPUT_SER_PATH --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Load .ser file from Canal output and calculate correlation between base pairs of the corresponding sequence.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_ser_path INPUT_SER_PATH
Path to ser file with inputs. Accepted formats: ser.
--output_csv_path OUTPUT_CSV_PATH
Path to output file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output plot. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_ser_path (string): Path to .ser file with data for single helical parameter. File type: input. Sample file. Accepted formats: SER
output_csv_path (string): Path to directory where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
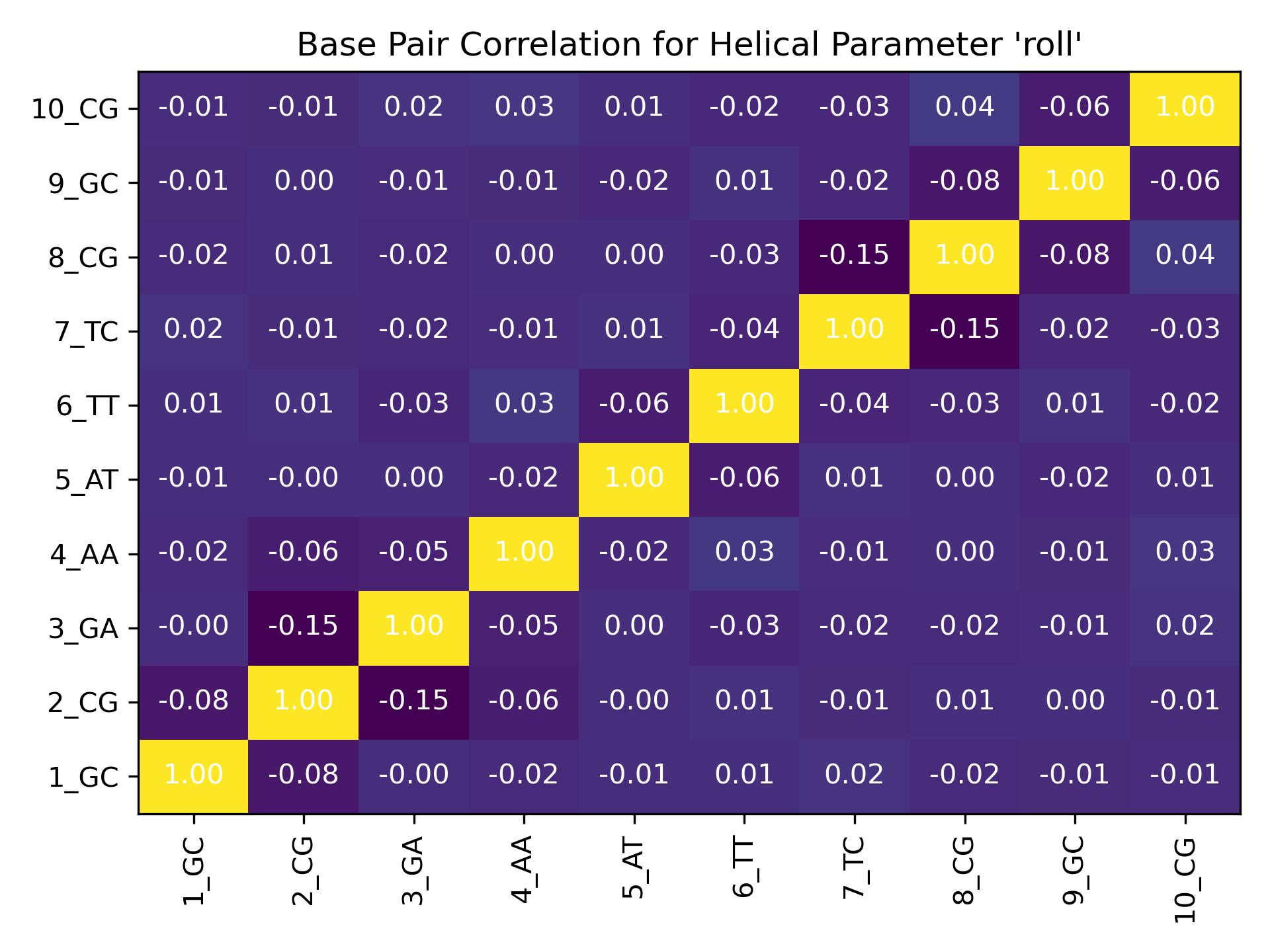{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
sequence (string): (None) Nucleic acid sequence for the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option)..
helpar_name (string): (None) helical parameter name to add to plot title..
seqpos (array): (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
sequence: CGCGAATTCGCG
Command line
interseqcorr --config config_interseqcorr.yml --input_ser_path canal_output_roll.ser --output_csv_path inter_seqcorr_roll.csv --output_jpg_path inter_seqcorr_roll.jpg
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG"
}
}
Command line
interseqcorr --config config_interseqcorr.json --input_ser_path canal_output_roll.ser --output_csv_path inter_seqcorr_roll.csv --output_jpg_path inter_seqcorr_roll.jpg
Interhpcorr
Calculate correlation between helical parameters for a single inter-base pair.
Get help
Command:
interhpcorr -h
usage: interhpcorr [-h] [--config CONFIG] --input_filename_shift INPUT_FILENAME_SHIFT --input_filename_slide INPUT_FILENAME_SLIDE --input_filename_rise INPUT_FILENAME_RISE --input_filename_tilt INPUT_FILENAME_TILT --input_filename_roll INPUT_FILENAME_ROLL --input_filename_twist INPUT_FILENAME_TWIST --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Load helical parameter file and save base data individually.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_filename_shift INPUT_FILENAME_SHIFT
Path to csv file with inputs. Accepted formats: csv.
--input_filename_slide INPUT_FILENAME_SLIDE
Path to csv file with inputs. Accepted formats: csv.
--input_filename_rise INPUT_FILENAME_RISE
Path to csv file with inputs. Accepted formats: csv.
--input_filename_tilt INPUT_FILENAME_TILT
Path to csv file with inputs. Accepted formats: csv.
--input_filename_roll INPUT_FILENAME_ROLL
Path to csv file with inputs. Accepted formats: csv.
--input_filename_twist INPUT_FILENAME_TWIST
Path to csv file with inputs. Accepted formats: csv.
--output_csv_path OUTPUT_CSV_PATH
Path to output file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output file. Accepted formats: csv.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_filename_shift (string): Path to .csv file with data for helical parameter ‘shift’. File type: input. Sample file. Accepted formats: CSV
input_filename_slide (string): Path to .csv file with data for helical parameter ‘slide’. File type: input. Sample file. Accepted formats: CSV
input_filename_rise (string): Path to .csv file with data for helical parameter ‘rise’. File type: input. Sample file. Accepted formats: CSV
input_filename_tilt (string): Path to .csv file with data for helical parameter ‘tilt’. File type: input. Sample file. Accepted formats: CSV
input_filename_roll (string): Path to .csv file with data for helical parameter ‘roll’. File type: input. Sample file. Accepted formats: CSV
input_filename_twist (string): Path to .csv file with data for helical parameter ‘twist’. File type: input. Sample file. Accepted formats: CSV
output_csv_path (string): Path to directory where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
basepair (string): (None) Name of basepair analyzed..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
remove_tmp: false
Command line
interhpcorr --config config_interhpcorr.yml --input_filename_shift series_shift_AA.csv --input_filename_slide series_slide_AA.csv --input_filename_rise series_rise_AA.csv --input_filename_tilt series_tilt_AA.csv --input_filename_roll series_roll_AA.csv --input_filename_twist series_twist_AA.csv --output_csv_path inter_hpcorr_ref.csv --output_jpg_path inter_hpcorr_ref.jpg
JSON
Common config file
{
"properties": {
"remove_tmp": false
}
}
Command line
interhpcorr --config config_interhpcorr.json --input_filename_shift series_shift_AA.csv --input_filename_slide series_slide_AA.csv --input_filename_rise series_rise_AA.csv --input_filename_tilt series_tilt_AA.csv --input_filename_roll series_roll_AA.csv --input_filename_twist series_twist_AA.csv --output_csv_path inter_hpcorr_ref.csv --output_jpg_path inter_hpcorr_ref.jpg
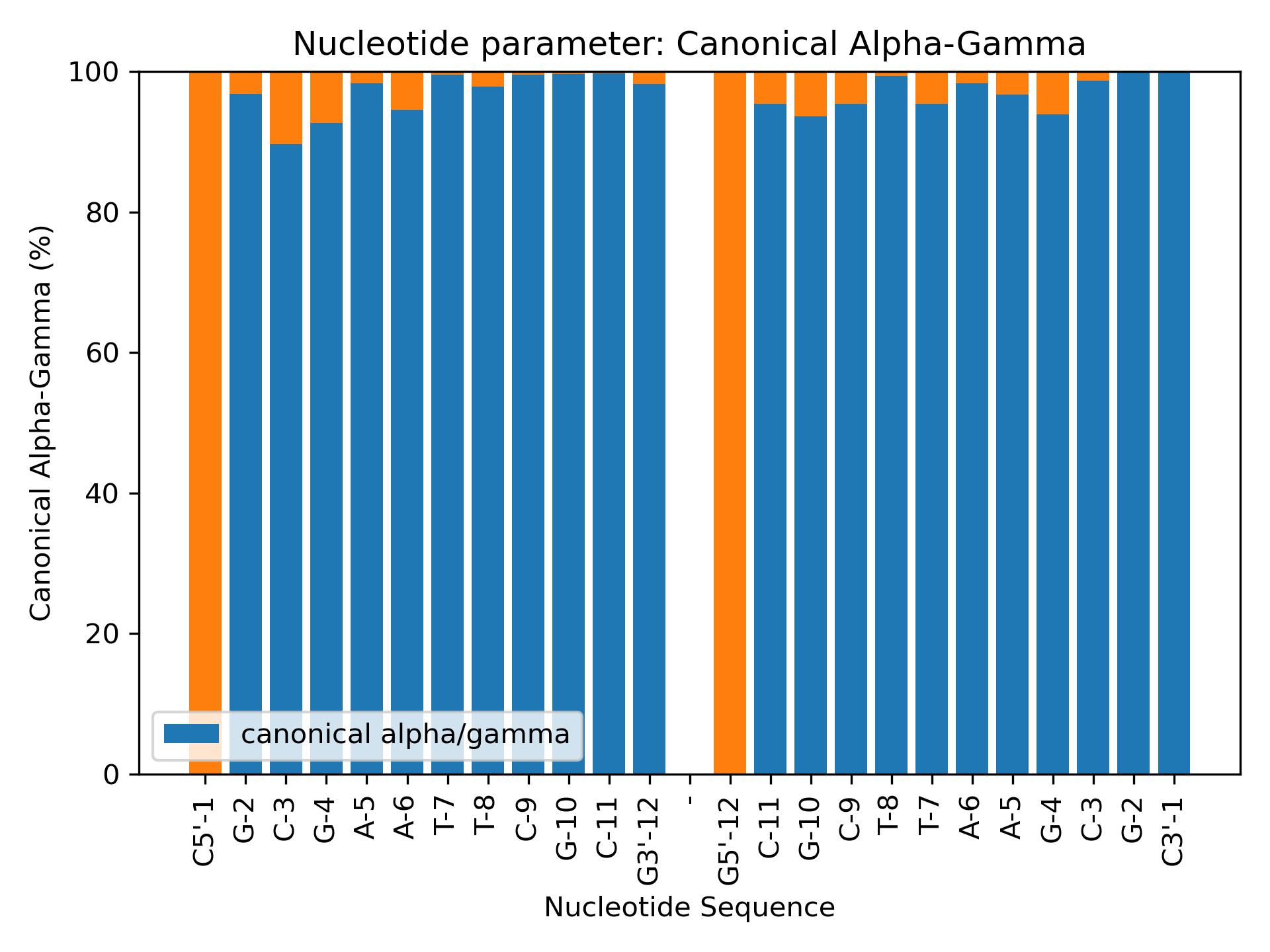
Canonicalag
Calculate Canonical Alpha/Gamma populations from alpha and gamma parameters.
Get help
Command:
canonicalag -h
usage: canonicalag [-h] [--config CONFIG] --input_alphaC_path INPUT_ALPHAC_PATH --input_alphaW_path INPUT_ALPHAW_PATH --input_gammaC_path INPUT_GAMMAC_PATH --input_gammaW_path INPUT_GAMMAW_PATH --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Calculate Canonical Alpha/Gamma distributions.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_alphaC_path INPUT_ALPHAC_PATH
Helical parameter <alphaC> input ser file path. Accepted formats: ser.
--input_alphaW_path INPUT_ALPHAW_PATH
Helical parameter <alphaW> input ser file path. Accepted formats: ser.
--input_gammaC_path INPUT_GAMMAC_PATH
Helical parameter <gammaC> input ser file path. Accepted formats: ser.
--input_gammaW_path INPUT_GAMMAW_PATH
Helical parameter <gammaW> input ser file path. Accepted formats: ser.
--output_csv_path OUTPUT_CSV_PATH
Path to output csv file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output jpg file. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_alphaC_path (string): Path to .ser file for helical parameter ‘alphaC’. File type: input. File type: input. Sample file. Accepted formats: SER
input_alphaW_path (string): Path to .ser file for helical parameter ‘alphaW’. File type: input. File type: input. Sample file. Accepted formats: SER
input_gammaC_path (string): Path to .ser file for helical parameter ‘gammaC’. File type: input. File type: input. Sample file. Accepted formats: SER
input_gammaW_path (string): Path to .ser file for helical parameter ‘gammaW’. File type: input. File type: input. Sample file. Accepted formats: SER
output_csv_path (string): Path to .csv file where output is saved. File type: output. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. File type: output. Sample file. Accepted formats: JPG
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
sequence (string): (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option)..
seqpos (array): (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
sequence: CGCGAATTCGCG
Command line
canonicalag --config config_canonicalag.yml --input_alphaC_path canal_output_alphaC.ser --input_alphaW_path canal_output_alphaW.ser --input_gammaC_path canal_output_gammaC.ser --input_gammaW_path canal_output_gammaW.ser --output_csv_path canonag_ref.csv --output_jpg_path canonag_ref.jpg
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG"
}
}
Command line
canonicalag --config config_canonicalag.json --input_alphaC_path canal_output_alphaC.ser --input_alphaW_path canal_output_alphaW.ser --input_gammaC_path canal_output_gammaC.ser --input_gammaW_path canal_output_gammaW.ser --output_csv_path canonag_ref.csv --output_jpg_path canonag_ref.jpg
Average_stiffness
Calculate average stiffness constants for each base pair of a trajectory’s series.
Get help
Command:
average_stiffness -h
usage: average_stiffness [-h] [--config CONFIG] --input_ser_path INPUT_SER_PATH --output_csv_path OUTPUT_CSV_PATH --output_jpg_path OUTPUT_JPG_PATH
Calculate average stiffness constants for each base pair of a trajectory's series.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
required arguments:
--input_ser_path INPUT_SER_PATH
Helical parameter input ser file path. Accepted formats: ser.
--output_csv_path OUTPUT_CSV_PATH
Path to output csv file. Accepted formats: csv.
--output_jpg_path OUTPUT_JPG_PATH
Path to output jpg file. Accepted formats: jpg.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_ser_path (string): Path to .ser file for helical parameter. File is expected to be a table, with the first column being an index and the rest the helical parameter values for each base/basepair. File type: input. Sample file. Accepted formats: SER
output_csv_path (string): Path to .csv file where output is saved. File type: output. Sample file. Accepted formats: CSV
output_jpg_path (string): Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: JPG
{kind=link}
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
KT (number): (0.592186827) Value of Boltzmann temperature factor..
sequence (string): (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the usecols option)..
helpar_name (string): (None) helical parameter name..
seqpos (array): (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
sequence: CGCGAATTCGCG
Command line
average_stiffness --config config_average_stiffness.yml --input_ser_path canal_output_roll.ser --output_csv_path stiffavg_roll.csv --output_jpg_path stiffavg_roll.jpg
JSON
Common config file
{
"properties": {
"sequence": "CGCGAATTCGCG"
}
}
Command line
average_stiffness --config config_average_stiffness.json --input_ser_path canal_output_roll.ser --output_csv_path stiffavg_roll.csv --output_jpg_path stiffavg_roll.jpg
Biobb_curves
Wrapper for the Cur+ executable that is part of the Curves+ software suite.
Get help
Command:
biobb_curves -h
usage: biobb_curves [-h] [--config CONFIG] --input_struc_path INPUT_STRUC_PATH --output_cda_path OUTPUT_CDA_PATH --output_lis_path OUTPUT_LIS_PATH [--input_top_path INPUT_TOP_PATH] [--output_zip_path OUTPUT_ZIP_PATH]
Execute Cur+ form the Curves+ software suite.
optional arguments:
-h, --help show this help message and exit
--config CONFIG Configuration file
--input_top_path INPUT_TOP_PATH
Topology file, needed along with .trj file (optional). Accepted formats: top.
--output_zip_path OUTPUT_ZIP_PATH
Filename to give to output files (except .cda and .lis files). Accepted formats: str.
required arguments:
--input_struc_path INPUT_STRUC_PATH
Trajectory or PDB input file. Accepted formats: trj, pdb.
--output_cda_path OUTPUT_CDA_PATH
Filename to give to output .cda file. Accepted formats: str.
--output_lis_path OUTPUT_LIS_PATH
Filename to give to output .lis file. Accepted formats: str.
I / O Arguments
Syntax: input_argument (datatype) : Definition
Config input / output arguments for this building block:
input_struc_path (string): Trajectory or PDB input file. File type: input. Sample file. Accepted formats: TRJ, PDB, NETCDF, NC
input_top_path (string): Topology file, needed along with .trj file (optional). File type: input. Sample file. Accepted formats: TOP, PDB
output_cda_path (string): Filename for Curves+ output .cda file. File type: output. Sample file. Accepted formats: CDA
output_lis_path (string): Filename for Curves+ output .lis file. File type: output. Sample file. Accepted formats: LIS
output_zip_path (string): Filename for .zip files containing Curves+ output that is not .cda or .lis files. File type: output. Sample file. Accepted formats: ZIP
Config
Syntax: input_parameter (datatype) - (default_value) Definition
Config parameters for this building block:
s1range (string): (None) Range of first strand. Must be specified in the form “start:end”..
s2range (string): (None) Range of second strand. Must be specified in the form “start:end”..
stdlib_path (string): (standard) Path to Curves’ standard library files for nucleotides. If not specified will look for ‘standard’ files in current directory..
itst (integer): (0) Iteration start index..
itnd (integer): (0) Iteration end index..
itdel (integer): (1) Iteration delimiter..
ions (boolean): (False) If True, helicoidal analysis of ions (or solvent molecules) around solute is carried out..
test (boolean): (False) If True, provide addition output in .lis file on fitting and axis generation..
line (boolean): (False) if True, find the best linear helical axis..
fit (boolean): (True) if True, fit a standard bases to the input coordinates (important for MD snapshots to avoid base distortions leading to noisy helical parameters)..
axfrm (boolean): (False) if True, generates closely spaced helical axis frames as input for Canal and Canion..
binary_path (string): (Cur+) Path to Curves+ executable, otherwise the program wil look for Cur+ executable in the binaries folder..
remove_tmp (boolean): (True) Remove temporal files..
restart (boolean): (False) Do not execute if output files exist..
sandbox_path (string): (./) Parent path to the sandbox directory..
YAML
Common config file
properties:
ions: true
s1range: '1:12'
Command line
biobb_curves --config config_biobb_curves.yml --input_struc_path structure.stripped.trj --input_top_path structure.stripped.top --output_cda_path curves_trj_output.cda --output_lis_path curves_trj_output.lis --output_zip_path curves_trj_output.zip
JSON
Common config file
{
"properties": {
"s1range": "1:12",
"ions": true
}
}
Command line
biobb_curves --config config_biobb_curves.json --input_struc_path structure.stripped.trj --input_top_path structure.stripped.top --output_cda_path curves_trj_output.cda --output_lis_path curves_trj_output.lis --output_zip_path curves_trj_output.zip