backbone package
Submodules
backbone.bipopulations module
Module containing the BIPopulations class and the command line interface.
- class backbone.bipopulations.BIPopulations(input_epsilC_path, input_epsilW_path, input_zetaC_path, input_zetaW_path, output_csv_path, output_jpg_path, properties=None, **kwargs)[source]
Bases:
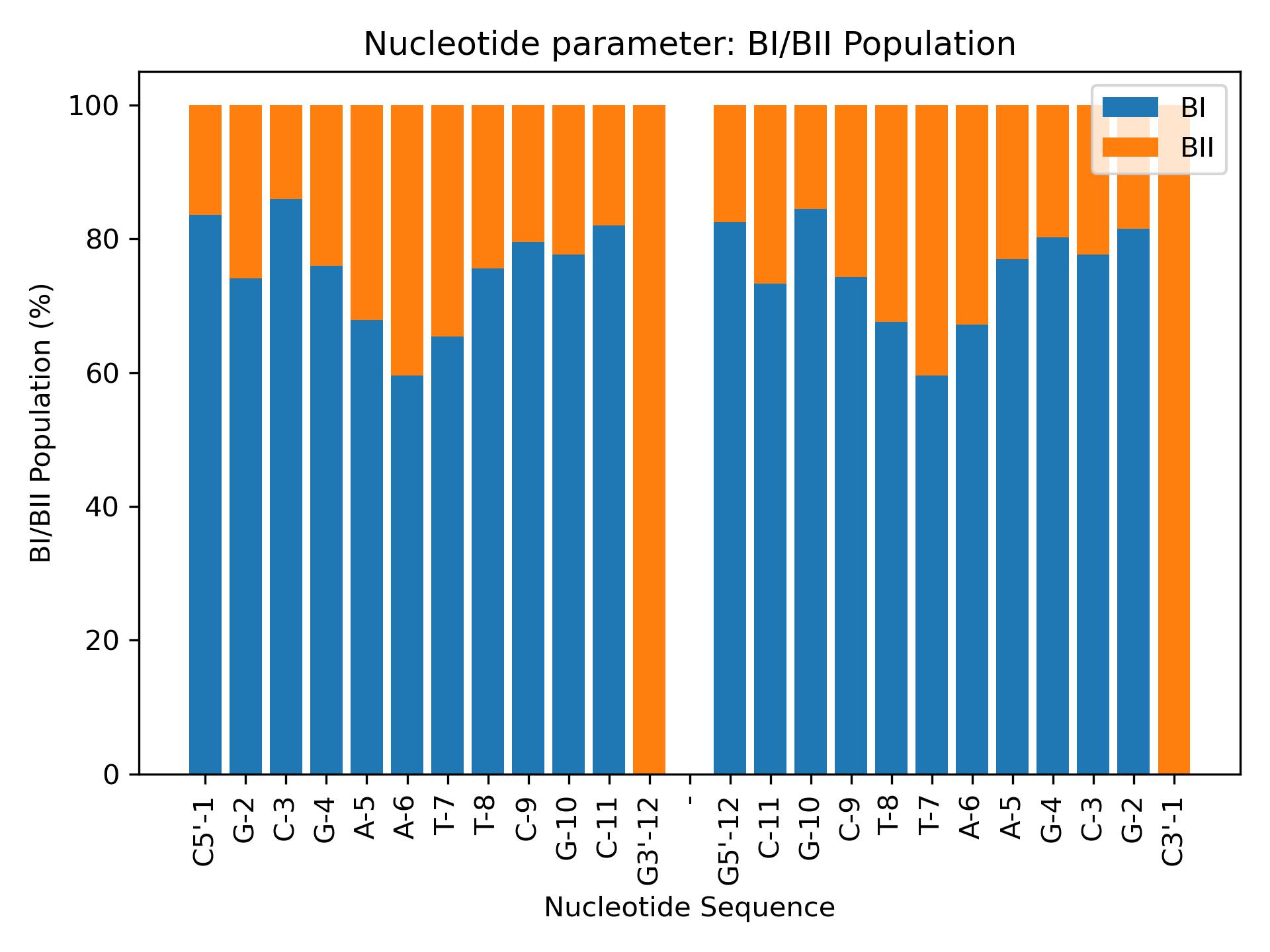
BiobbObjectbiobb_dna BIPopulationsCalculate BI/BII populations from epsilon and zeta parameters.Calculate BI/BII populations from epsilon and zeta parameters.- Parameters:
input_epsilC_path (str) – Path to .ser file for helical parameter ‘epsilC’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_epsilW_path (str) –
Path to .ser file for helical parameter ‘epsilW’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_zetaC_path (str) –
Path to .ser file for helical parameter ‘zetaC’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_zetaW_path (str) –
Path to .ser file for helical parameter ‘zetaW’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to .csv file where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.backbone.bipopulations import bipopulations prop = { 'sequence': 'GCAT', } bipopulations( input_epsilC_path='/path/to/epsilC.ser', input_epsilW_path='/path/to/epsilW.ser', input_zetaC_path='/path/to/zetaC.ser', input_zetaW_path='/path/to/zetaW.ser', output_csv_path='/path/to/table/output.csv', output_jpg_path='/path/to/table/output.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
- launch() int[source]
Execute the
BIPopulationsobject.
{kind=link}
- backbone.bipopulations.bipopulations(input_epsilC_path: str, input_epsilW_path: str, input_zetaC_path: str, input_zetaW_path: str, output_csv_path: str, output_jpg_path: str, properties: dict | None = None, **kwargs) int[source]
- biobb_dna BIPopulationsCalculate BI/BII populations from epsilon and zeta parameters.Calculate BI/BII populations from epsilon and zeta parameters.
- Parameters:
input_epsilC_path (str) –
Path to .ser file for helical parameter ‘epsilC’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_epsilW_path (str) –
Path to .ser file for helical parameter ‘epsilW’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_zetaC_path (str) –
Path to .ser file for helical parameter ‘zetaC’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_zetaW_path (str) –
Path to .ser file for helical parameter ‘zetaW’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to .csv file where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.backbone.bipopulations import bipopulations prop = { 'sequence': 'GCAT', } bipopulations( input_epsilC_path='/path/to/epsilC.ser', input_epsilW_path='/path/to/epsilW.ser', input_zetaC_path='/path/to/zetaC.ser', input_zetaW_path='/path/to/zetaW.ser', output_csv_path='/path/to/table/output.csv', output_jpg_path='/path/to/table/output.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
backbone.canonicalag module
Module containing the CanonicalAG class and the command line interface.
- class backbone.canonicalag.CanonicalAG(input_alphaC_path, input_alphaW_path, input_gammaC_path, input_gammaW_path, output_csv_path, output_jpg_path, properties=None, **kwargs)[source]
Bases:
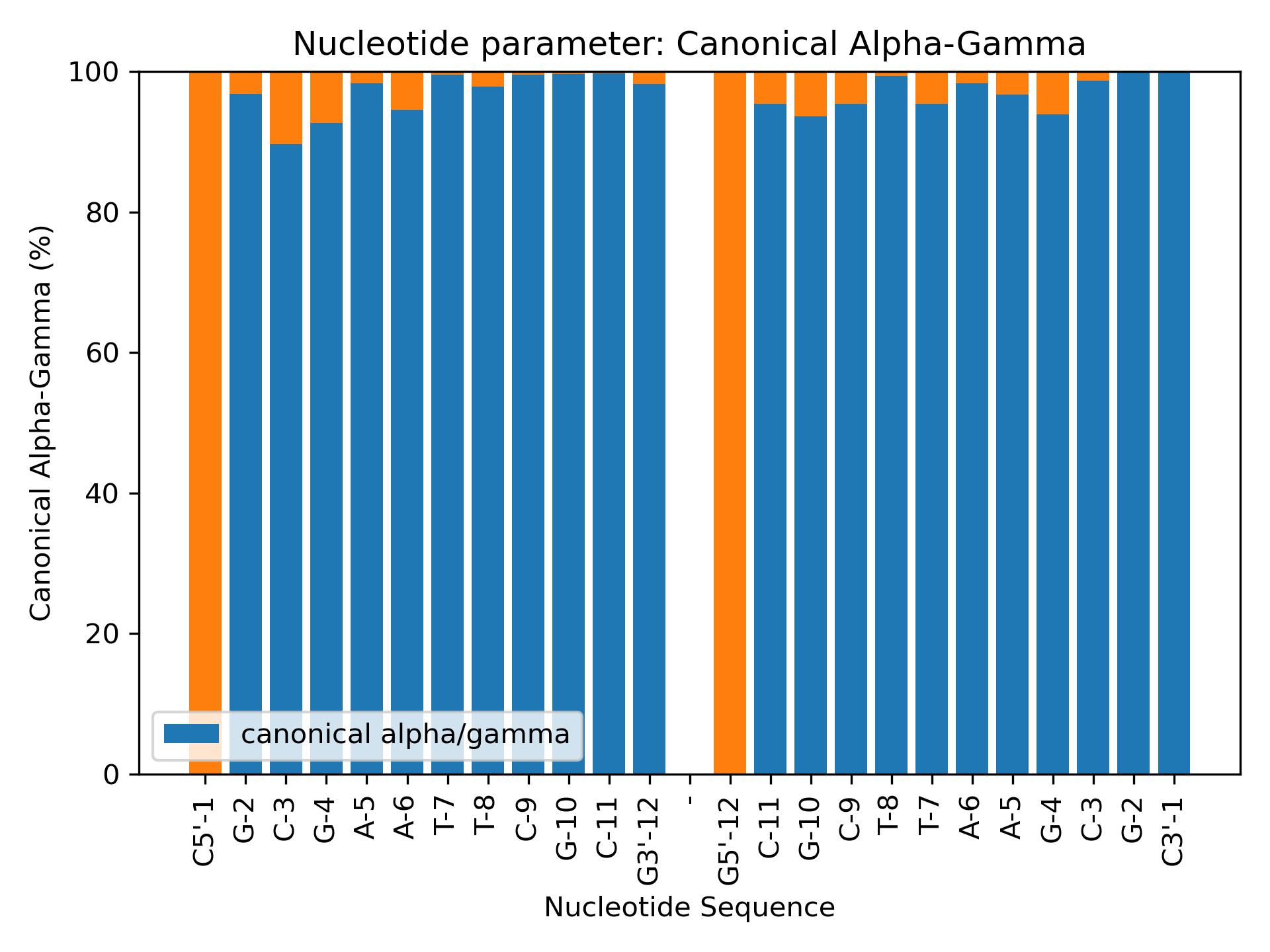
BiobbObjectbiobb_dna CanonicalAGCalculate Canonical Alpha/Gamma populations from alpha and gamma parameters.Calculate Canonical Alpha/Gamma populations from alpha and gamma parameters.- Parameters:
input_alphaC_path (str) –
Path to .ser file for helical parameter ‘alphaC’. File type: input. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_alphaW_path (str) –
Path to .ser file for helical parameter ‘alphaW’. File type: input. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_gammaC_path (str) –
Path to .ser file for helical parameter ‘gammaC’. File type: input. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_gammaW_path (str) –
Path to .ser file for helical parameter ‘gammaW’. File type: input. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to .csv file where output is saved. File type: output. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.backbone.canonicalag import canonicalag prop = { 'seqpos': [1,2], 'sequence': 'GCAT', } canonicalag( input_alphaC_path='/path/to/alphaC.ser', input_alphaW_path='/path/to/alphaW.ser', input_gammaC_path='/path/to/gammaC.ser', input_gammaW_path='/path/to/gammaW.ser', output_csv_path='/path/to/table/output.csv', output_jpg_path='/path/to/table/output.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
- launch() int[source]
Execute the
CanonicalAGobject.
{kind=link}
- backbone.canonicalag.canonicalag(input_alphaC_path: str, input_alphaW_path: str, input_gammaC_path: str, input_gammaW_path: str, output_csv_path: str, output_jpg_path: str, properties: dict | None = None, **kwargs) int[source]
- biobb_dna CanonicalAGCalculate Canonical Alpha/Gamma populations from alpha and gamma parameters.Calculate Canonical Alpha/Gamma populations from alpha and gamma parameters.
- Parameters:
input_alphaC_path (str) –
Path to .ser file for helical parameter ‘alphaC’. File type: input. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_alphaW_path (str) –
Path to .ser file for helical parameter ‘alphaW’. File type: input. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_gammaC_path (str) –
Path to .ser file for helical parameter ‘gammaC’. File type: input. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_gammaW_path (str) –
Path to .ser file for helical parameter ‘gammaW’. File type: input. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to .csv file where output is saved. File type: output. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.backbone.canonicalag import canonicalag prop = { 'seqpos': [1,2], 'sequence': 'GCAT', } canonicalag( input_alphaC_path='/path/to/alphaC.ser', input_alphaW_path='/path/to/alphaW.ser', input_gammaC_path='/path/to/gammaC.ser', input_gammaW_path='/path/to/gammaW.ser', output_csv_path='/path/to/table/output.csv', output_jpg_path='/path/to/table/output.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
backbone.puckering module
Module containing the Puckering class and the command line interface.
- class backbone.puckering.Puckering(input_phaseC_path, input_phaseW_path, output_csv_path, output_jpg_path, properties=None, **kwargs)[source]
Bases:
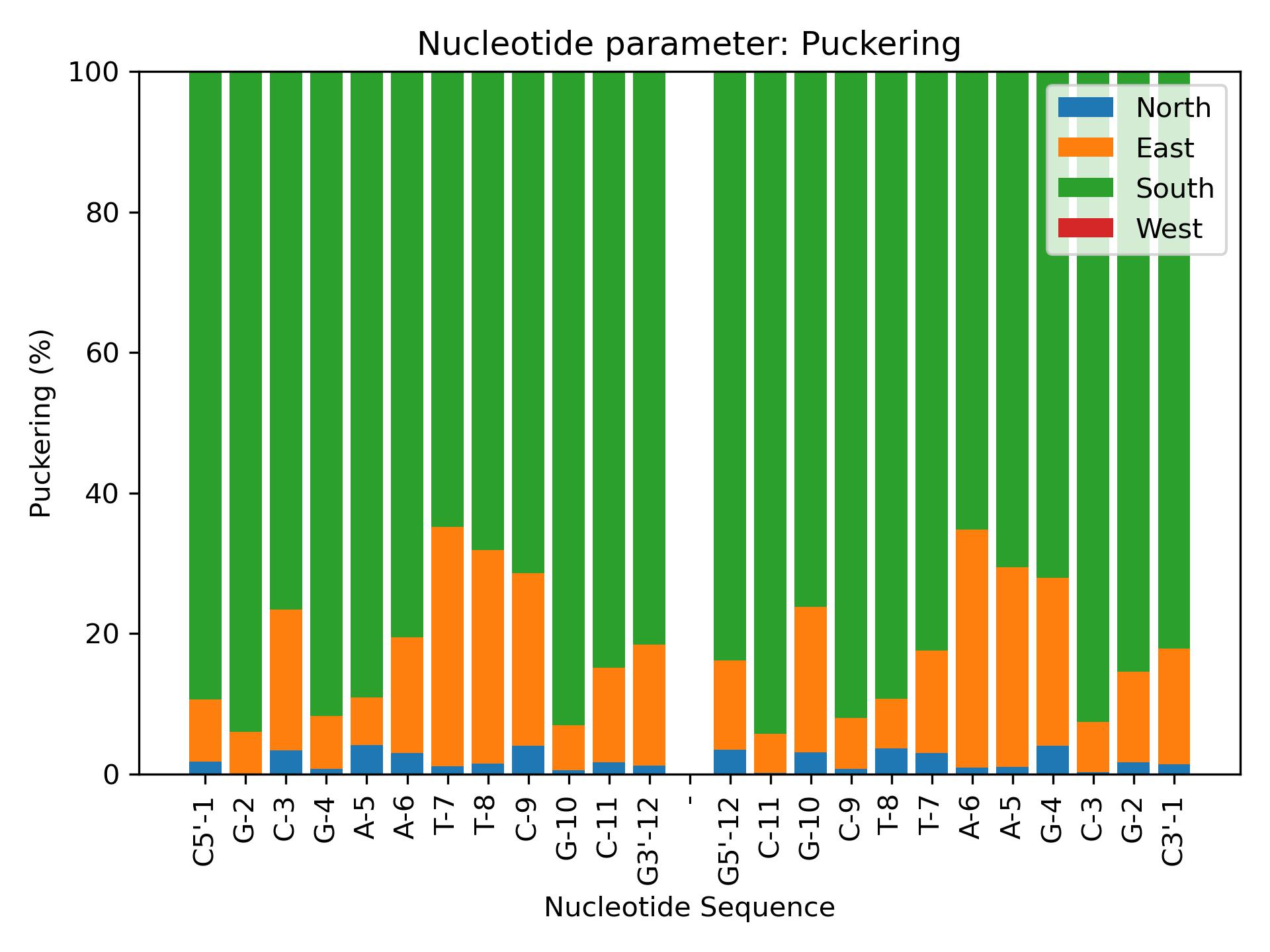
BiobbObjectbiobb_dna PuckeringCalculate Puckering from phase parameters.Calculate North/East/West/South distribution of sugar puckering backbone torsions.- Parameters:
input_phaseC_path (str) –
Path to .ser file for helical parameter ‘phaseC’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_phaseW_path (str) –
Path to .ser file for helical parameter ‘phaseW’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to .csv file where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
stride (int) - (1000) granularity of the number of snapshots for plotting time series.
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.backbone.puckering import puckering prop = { 'sequence': 'GCAT', } puckering( input_phaseC_path='/path/to/phaseC.ser', input_phaseW_path='/path/to/phaseW.ser', output_csv_path='/path/to/table/output.csv', output_jpg_path='/path/to/table/output.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
{kind=link}
- backbone.puckering.puckering(input_phaseC_path: str, input_phaseW_path: str, output_csv_path: str, output_jpg_path: str, properties: dict | None = None, **kwargs) int[source]
- biobb_dna PuckeringCalculate Puckering from phase parameters.Calculate North/East/West/South distribution of sugar puckering backbone torsions.
- Parameters:
input_phaseC_path (str) –
Path to .ser file for helical parameter ‘phaseC’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_phaseW_path (str) –
Path to .ser file for helical parameter ‘phaseW’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to .csv file where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence corresponding to the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
stride (int) - (1000) granularity of the number of snapshots for plotting time series.
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.backbone.puckering import puckering prop = { 'sequence': 'GCAT', } puckering( input_phaseC_path='/path/to/phaseC.ser', input_phaseW_path='/path/to/phaseW.ser', output_csv_path='/path/to/table/output.csv', output_jpg_path='/path/to/table/output.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl