intrabp_correlations package
Correlation functions for intra-base pairs.
Submodules
intrabp_correlations.intrahpcorr module
Module containing the IntraHelParCorrelation class and the command line interface.
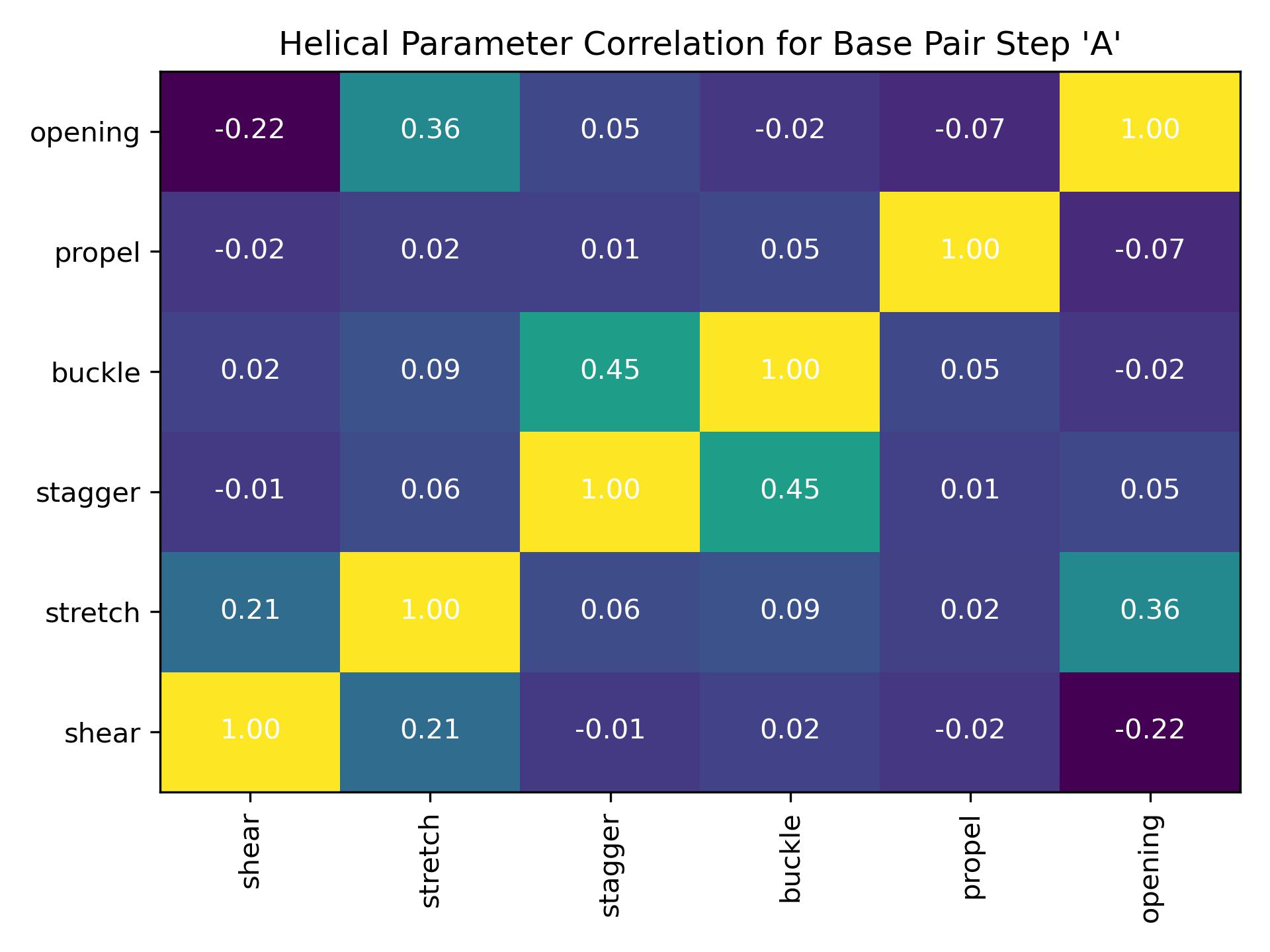
- class intrabp_correlations.intrahpcorr.IntraHelParCorrelation(input_filename_shear, input_filename_stretch, input_filename_stagger, input_filename_buckle, input_filename_propel, input_filename_opening, output_csv_path, output_jpg_path, properties=None, **kwargs)[source]
Bases:
BiobbObjectbiobb_dna IntraHelParCorrelationCalculate correlation between helical parameters for a single intra-base pair.Calculate correlation between helical parameters for a single intra-base pair.- Parameters:
input_filename_shear (str) – Path to .csv file with data for helical parameter ‘shear’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_stretch (str) –
Path to .csv file with data for helical parameter ‘stretch’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_stagger (str) –
Path to .csv file with data for helical parameter ‘stagger’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_buckle (str) –
Path to .csv file with data for helical parameter ‘buckle’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_propel (str) –
Path to .csv file with data for helical parameter ‘propeller’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_opening (str) –
Path to .csv file with data for helical parameter ‘opening’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
output_csv_path (str) –
Path to directory where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
base (str) - (None) Name of base analyzed.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.intrabp_correlations.intrahpcorr import intrahpcorr prop = { 'base': 'A', } intrahpcorr( input_filename_shear='path/to/shear.csv', input_filename_stretch='path/to/stretch.csv', input_filename_stagger='path/to/stagger.csv', input_filename_buckle='path/to/buckle.csv', input_filename_propel='path/to/propel.csv', input_filename_opening='path/to/opening.csv', output_csv_path='path/to/output/file.csv', output_jpg_path='path/to/output/file.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
- launch() int[source]
Execute the
IntraHelParCorrelationobject.
{kind=link}
- intrabp_correlations.intrahpcorr.intrahpcorr(input_filename_shear: str, input_filename_stretch: str, input_filename_stagger: str, input_filename_buckle: str, input_filename_propel: str, input_filename_opening: str, output_csv_path: str, output_jpg_path: str, properties: dict | None = None, **kwargs) int[source]
- biobb_dna IntraHelParCorrelationCalculate correlation between helical parameters for a single intra-base pair.Calculate correlation between helical parameters for a single intra-base pair.
- Parameters:
input_filename_shear (str) –
Path to .csv file with data for helical parameter ‘shear’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_stretch (str) –
Path to .csv file with data for helical parameter ‘stretch’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_stagger (str) –
Path to .csv file with data for helical parameter ‘stagger’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_buckle (str) –
Path to .csv file with data for helical parameter ‘buckle’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_propel (str) –
Path to .csv file with data for helical parameter ‘propeller’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
input_filename_opening (str) –
Path to .csv file with data for helical parameter ‘opening’. File type: input. Sample file. Accepted formats: csv (edam:format_3752).
output_csv_path (str) –
Path to directory where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
base (str) - (None) Name of base analyzed.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.intrabp_correlations.intrahpcorr import intrahpcorr prop = { 'base': 'A', } intrahpcorr( input_filename_shear='path/to/shear.csv', input_filename_stretch='path/to/stretch.csv', input_filename_stagger='path/to/stagger.csv', input_filename_buckle='path/to/buckle.csv', input_filename_propel='path/to/propel.csv', input_filename_opening='path/to/opening.csv', output_csv_path='path/to/output/file.csv', output_jpg_path='path/to/output/file.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
intrabp_correlations.intraseqcorr
Module containing the IntraSequenceCorrelation class and the command line interface.
- class intrabp_correlations.intraseqcorr.IntraSequenceCorrelation(input_ser_path, output_csv_path, output_jpg_path, properties=None, **kwargs)[source]
Bases:
BiobbObjectbiobb_dna IntraSequenceCorrelationCalculate correlation between all intra-base pairs of a single sequence and for a single helical parameter.Calculate correlation between all intra-base pairs of a single sequence and for a single helical parameter.- Parameters:
input_ser_path (str) –
Path to .ser file with data for single helical parameter. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to directory where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence for the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
helpar_name (str) - (None) helical parameter name to add to plot title.
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.intrabp_correlations.intraseqcorr import intraseqcorr intraseqcorr( input_ser_path='path/to/input/file.ser', output_csv_path='path/to/output/file.csv', output_jpg_path='path/to/output/plot.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
- launch() int[source]
Execute the
HelParCorrelationobject.
{kind=link}
- intrabp_correlations.intraseqcorr.intraseqcorr(input_ser_path: str, output_csv_path: str, output_jpg_path: str, properties: dict | None = None, **kwargs) int[source]
- biobb_dna IntraSequenceCorrelationCalculate correlation between all intra-base pairs of a single sequence and for a single helical parameter.Calculate correlation between all intra-base pairs of a single sequence and for a single helical parameter.
- Parameters:
input_ser_path (str) –
Path to .ser file with data for single helical parameter. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to directory where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence for the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
helpar_name (str) - (None) helical parameter name to add to plot title.
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.intrabp_correlations.intraseqcorr import intraseqcorr intraseqcorr( input_ser_path='path/to/input/file.ser', output_csv_path='path/to/output/file.csv', output_jpg_path='path/to/output/plot.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
intrabp_correlations.intrabpcorr module
Module containing the IntraBasePairCorrelation class and the command line interface.
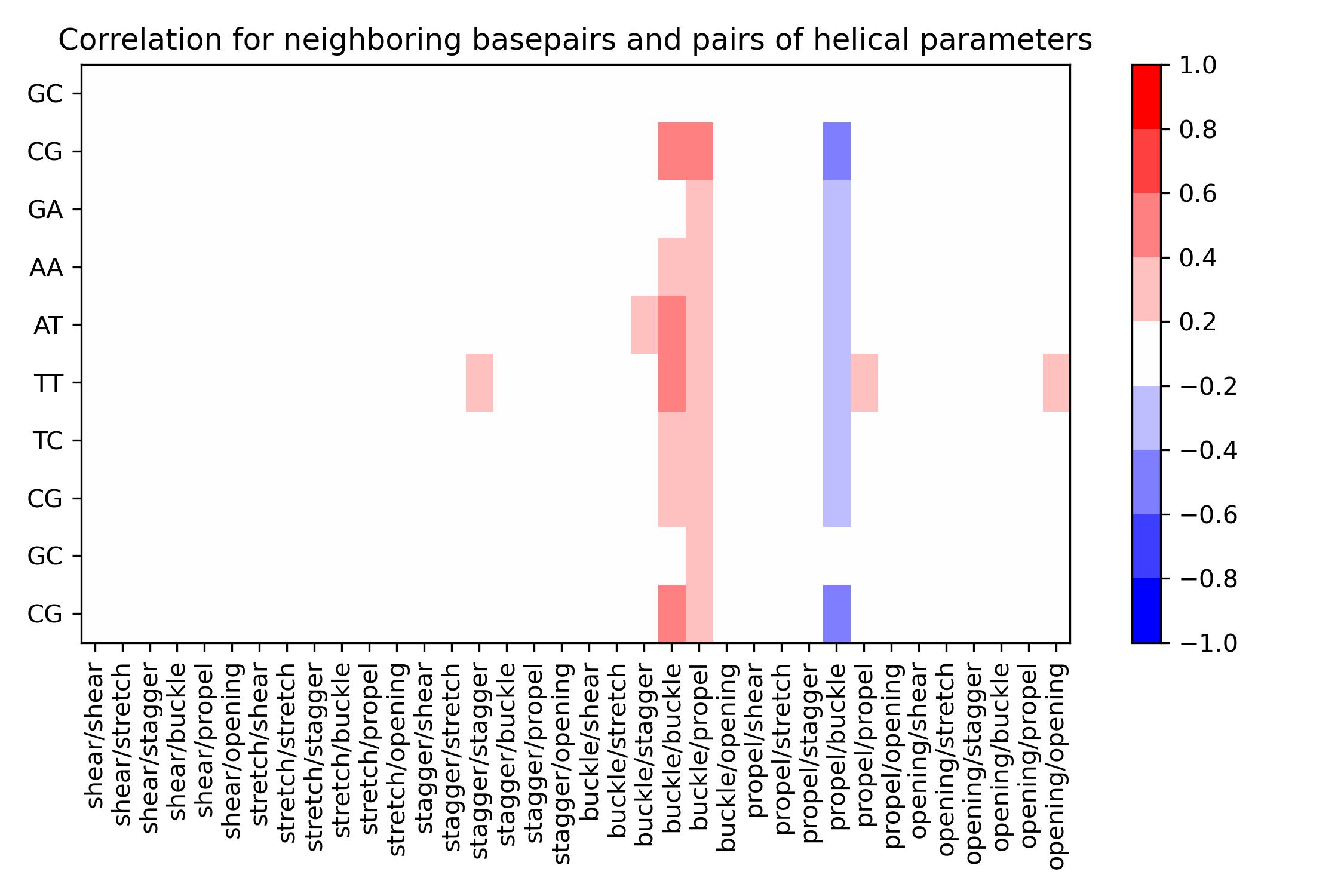
- class intrabp_correlations.intrabpcorr.IntraBasePairCorrelation(input_filename_shear, input_filename_stretch, input_filename_stagger, input_filename_buckle, input_filename_propel, input_filename_opening, output_csv_path, output_jpg_path, properties=None, **kwargs)[source]
Bases:
BiobbObjectbiobb_dna IntraBasePairCorrelationCalculate correlation between all intra-base pairs of a single sequence and for a single helical parameter.Calculate correlation between neighboring base pairs and pairs of helical parameters.- Parameters:
input_filename_shear (str) –
Path to .ser file with data for helical parameter ‘shear’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_stretch (str) –
Path to .ser file with data for helical parameter ‘stretch’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_stagger (str) –
Path to .ser file with data for helical parameter ‘stagger’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_buckle (str) –
Path to .ser file with data for helical parameter ‘buckle’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_propel (str) –
Path to .ser file with data for helical parameter ‘propel’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_opening (str) –
Path to .ser file with data for helical parameter ‘opening’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to directory where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence for the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.intrabp_correlations.intrabpcorr import intrabpcorr intrabpcorr( input_filename_shear='path/to/input/shear.ser', input_filename_stretch='path/to/input/stretch.ser', input_filename_stagger='path/to/input/stagger.ser', input_filename_buckle='path/to/input/buckle.ser', input_filename_propel='path/to/input/propel.ser', input_filename_opening='path/to/input/opening.ser', output_csv_path='path/to/output/file.csv', output_jpg_path='path/to/output/plot.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl
- launch() int[source]
Execute the
HelParCorrelationobject.
{kind=link}
- intrabp_correlations.intrabpcorr.intrabpcorr(input_filename_shear: str, input_filename_stretch: str, input_filename_stagger: str, input_filename_buckle: str, input_filename_propel: str, input_filename_opening: str, output_csv_path: str, output_jpg_path: str, properties: dict | None = None, **kwargs) int[source]
- biobb_dna IntraBasePairCorrelationCalculate correlation between all intra-base pairs of a single sequence and for a single helical parameter.Calculate correlation between neighboring base pairs and pairs of helical parameters.
- Parameters:
input_filename_shear (str) –
Path to .ser file with data for helical parameter ‘shear’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_stretch (str) –
Path to .ser file with data for helical parameter ‘stretch’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_stagger (str) –
Path to .ser file with data for helical parameter ‘stagger’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_buckle (str) –
Path to .ser file with data for helical parameter ‘buckle’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_propel (str) –
Path to .ser file with data for helical parameter ‘propel’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
input_filename_opening (str) –
Path to .ser file with data for helical parameter ‘opening’. File type: input. Sample file. Accepted formats: ser (edam:format_2330).
output_csv_path (str) –
Path to directory where output is saved. File type: output. Sample file. Accepted formats: csv (edam:format_3752).
output_jpg_path (str) –
Path to .jpg file where output is saved. File type: output. Sample file. Accepted formats: jpg (edam:format_3579).
properties (dict) –
sequence (str) - (None) Nucleic acid sequence for the input .ser file. Length of sequence is expected to be the same as the total number of columns in the .ser file, minus the index column (even if later on a subset of columns is selected with the seqpos option).
seqpos (list) - (None) list of sequence positions (columns indices starting by 0) to analyze. If not specified it will analyse the complete sequence.
remove_tmp (bool) - (True) [WF property] Remove temporal files.
restart (bool) - (False) [WF property] Do not execute if output files exist.
sandbox_path (str) - (“./”) [WF property] Parent path to the sandbox directory.
Examples
This is a use example of how to use the building block from Python:
from biobb_dna.intrabp_correlations.intrabpcorr import intrabpcorr intrabpcorr( input_filename_shear='path/to/input/shear.ser', input_filename_stretch='path/to/input/stretch.ser', input_filename_stagger='path/to/input/stagger.ser', input_filename_buckle='path/to/input/buckle.ser', input_filename_propel='path/to/input/propel.ser', input_filename_opening='path/to/input/opening.ser', output_csv_path='path/to/output/file.csv', output_jpg_path='path/to/output/plot.jpg', properties=prop)
- Info:
- wrapped_software:
name: In house
license: Apache-2.0
- ontology:
name: EDAM
schema: http://edamontology.org/EDAM.owl